ГЕМОФИЛИЯ
ОПРЕДЕЛЕНИЕ
Гемофилии — группа заболеваний, при которых дефицит факторов свёртывания (чаще VIII и IX) приводит к появлению геморрагического синдрома. Выделяют врождённые и приобретённые формы. Чаще обнаруживают врождённые гемофилии: гемофилия А (дефицит фактора VIII) и гемофилия В (дефицит фактора IX, — болезнь Кристмаса). Намного реже встречается гемофилия С (дефицит фактора XI), очень редко — конкомитированная гемофилия (одновременный дефицит факторов VIII и IX), часто сопровождаемая нарушением цветового зрения. Приобретённая гемофилия у детей бывает редко, как правило, при появлении антител к факторам свёртывания при аутоиммунных и миелопролиферативных заболеваниях.
ЭПИДЕМИОЛОГИЯ
Заболеваемость гемофилией составляет 13-14 на 100 000 мужчин. Соотношение гемофилии А и гемофилии В составляет 4:1. Наследственный характер гемофилии отмечен у 70-90% пациентов, спорадический — у 10-30%.
КЛАССИФИКАЦИЯ
• Очень тяжёлая форма гемофилии (при очень тяжёлой форме гемофилии активность фактора VIII/IX не превышает 0,99%).
• Тяжёлая форма гемофилии (активность фактора VIII/IX составляет 1-2,99%).
• Среднетяжёлая форма гемофилии (активность фактора VIII/ IX - 3-4%).
• Лёгкая форма гемофилии (активность фактора VIII/IX — 5-12%).
• Стёртая форма гемофилии (активность фактора VIII/IX — 13-50%).
ЭТИОЛОГИЯ
Наследование гемофилии Х-сцепленное. Все дочери больных гемофилией — облигатные носители аномальных генов; все сыновья — здоровы. Вероятность того, что сын матери-носительницы будет болен гемофилией, составляет 50%, и вероятность того, что её дочь станет носительницей заболевания, также 50% (рис. 18-1).

Рис. 18-1. Наследование гемофилии (схема).
Гемофилией могут страдать девочки, рождённые от больного гемофилией мужчины и женщины-носительницы, а также при синдроме Тёрнера. У женщин-носительниц кровоточивость может происходить во время менструаций и при операциях.
КЛИНИЧЕСКАЯ КАРТИНА
Клиническая картина гемофилии А и гемофилии В идентична; тип гемофилии устанавливают только при лабораторном исследовании, в том числе и при количественном определении факторов свёртывания.
Кровотечения
Кровотечения отсроченные, возникают через несколько часов после травмы. Кровотечения в неонатальном периоде: кефалогематома, кровоизлияния в области ягодиц при ягодичном предлежании, кровотечение из пупочного канатика. Позже — кровотечения при прорезывании зубов или при ранении уздечки языка, гематомы в местах ушибов и внутримышечных инъекций, кровотечение при обрезании крайней плоти.
Желудочно-кишечные кровотечения характерны для детей старшего возраста, они связаны с эрозивно-язвенной патологией ЖКТ.
Кровоизлияния
Возможны кровоизлияния в суставы, чаще в крупные: коленные, голеностопные, локтевые. С момента самостоятельного хождения ребёнка ведущим симптомом становятся межмышечные гематомы. Кровь в полости сустава вызывает воспаление синовиальной оболочки, а повторные кровоизлияния приводят к разрушению суставного хряща, развитию остеоартроза, фиброза и анкилоза сустава с последующей атрофией мышц. Пострадавший сустав обычно становится местом повторных кровоизлияний.
Кровоизлияние в подвздошно-поясничную мышцу вызывают боль в животе, сгибательную контрактуру бедра (клинически имитирует поражение тазобедренного сустава), ригидность мышц передней брюшной стенки, что нередко принимают за острый аппендицит. При пальпации области поражённой мышцы обнаруживают плотное болезненное образование.
Симптомы при кровоизлиянии в головной мозг развиваются стремительно или по нарастанию (беспокойство или заторможённость, рвота, головная боль, стволовые симптомы: горизонтальный и вертикальный нистагм, анизокория, расстройства ритма дыхания и сердечных сокращений). Любые признаки повышения внутричерепного давления при отсутствии в анамнезе травмы головы — показание к немедленному заместительному лечению. Кровоизлияния в спинной мозг встречаются реже; они вызывают нарастающие периферические парезы.
Гематурия
Гематурию чаще отмечают у детей старше 5 лет. Причинами её могут быть травмы поясничной области, иммунокомплексное поражение почек, высокая активность урокиназы, оксалурия у пациентов с повторными гемартрозами и частыми приёмами анальгетиков, аномалии развития или положения почек. Макрогематурия чаще возникает спонтанно. Иногда ей сопутствуют дизурия, боли в поясничной области (вплоть до почечной колики), по ходу мочеточников или уретры. После нескольких болезненных позывов к мочеиспусканию отходят сгустки крови и боли уменьшаются.
Лёгкая форма гемофилии сопровождается минимальной кровоточивостью, и её выявляют в более зрелом возрасте при оперативных вмешательствах или значительных травмах.
ДИАГНОСТИКА Лабораторная диагностика
• Диагностируют по удлинению времени свёртывания цельной крови и активированного частичного тромбопластинового времени (АЧТВ); время кровотечения и протромбиновое время не изменены.
• Тип и тяжесть гемофилии определяют по снижению коагулянтной активности антигемофильных глобулинов в плазме (факторы VIII и IX).
• Поскольку активность фактора VIII может быть снижена и при болезни Виллебранда, у пациентов с впервые обнаруженной гемофилией А следует определить содержание антигена фактора Виллебранда (при гемофилии А содержание антигена остаётся нормальным).
• Скрининг пациентов на наличие ингибиторов к факторам VIII и/или IX особенно необходим перед плановыми хирургическими вмешательствами.
• Пренатальная диагностика и выявление носителей.
Гемофилию А диагностируют, анализируя ген фактора VIII, а гемофилию В — ген фактора IX. Применяют два метода: «полимеразная цепная реакция — полиморфизм длин рестрикционных фрагментов» и «полимеразная цепная реакция с обратной транскрипцией». Для каждого метода необходимо небольшое количество крови или биоптата ворсинок хориона, что даёт возможность диагностировать гемофилию пренатально на ранней стадии беременности (8-12 нед).
ЛЕЧЕНИЕ
Основной компонент лечения гемофилии — своевременное адекватное заместительное воздействие, восполняющее уровень дефицитного фактора в плазме.
В настоящее время существует три метода лечения больных гемофилией:
• профилактический;
• лечение на дому;
• лечение по факту возникновения кровотечения.
Профилактический метод
Это наиболее прогрессивный метод. Его цель — поддержание активности дефицитного фактора на уровне около 5% нормы во избежание кровоизлияний в суставы. Профилактическое лечение начинают в возрасте 1-2 года до возникновения первого гемартроза или сразу после него. В лечении применяют концентраты факторов свёртывания (КФС) высокой степени очистки (табл. 18-1 и 18-2). Препараты вводят 3 раза в неделю при гемофилии А и 2 раза в неделю при гемофилии В (так как период полувыведения фактора IX более продолжительный) из расчёта 25-40 МЕ/кг. Длительность профилактического лечения - от нескольких месяцев до пожизненного. У пациентов отсутствуют поражения опорно-двигательного аппарата, они полностью социально адаптированы и могут заниматься спортом.
Таблица 18-1. Концентраты фактора свёртывания крови VIII, зарегистрированные в России
|
Препарат |
Способ получения |
Инактивация вируса |
Применение |
|
Гемофил М* |
Иммуноаффинная хроматография с моноклональными антителами к фактору VIII* |
Сольвент-детергентная + иммуноаффинная хроматография |
Гемофилия А, Ингибиторная гемофилия А** |
|
Иммунат* |
Ионообменная хроматография |
Двойная: сольвент- детергентная + термическая |
Гемофилия А, Ингибиторная гемофилия А,*‘ болезнь Виллебранда |
|
Коэйт-ДВИ* |
Хроматография |
Двойная: сольвент- детергентная + термическая |
Гемофилия А, ингибиторная гемофилия А** |
|
Эмоклот Д.И. * |
Хроматография |
Двойная: сольвент- детергентная + термическая |
Гемофилия А, ингибиторная гемофилия А ** |
* Стабилизирован альбумином. ** При титре ингибитора менее 10 БЕ (единиц Бетезда) на 1 мл.
Таблица. 18-2. Концентраты фактора свёртывания крови IX, зарегистрированные в России
|
Препарат |
Способ получения |
Инактивация вируса |
Применение |
|
Иммунин* |
Ионообменная хроматография |
Двойная: сольвент- детергентная + термическая |
Гемофилия В, ингибиторная гемофилия В |
|
Аимафикс* |
Хроматография |
Двойная: сольвент- детергентная + термическая |
Гемофилия В |
|
Октанайн Ф* |
Хроматография |
Двойная: сольвент- детергентная + термическая |
Гемофилия В, ингибиторная гемофилия В |
Подходы в назначении этих препаратов такие же, как и при гемофилии А.
Лечение на дому
Рекомендуется больным либо с менее выраженным геморрагическим синдромом, либо при ограниченных возможностях лекарственного обеспечения. Препарат вводят сразу после травмы или при малейших признаках начинающегося кровоизлияния. Немедленное введение препарата способствует остановке кровотечения на раннем этапе, предотвращает повреждение тканей или образование массивного гемартроза при меньшем расходе препаратов. Для лечения на дому также применяют КФС.
Лечение по факту возникновения кровотечения
Такое лечение требует небольшого количества препаратов, но не позволяет избежать массивных межмышечных и забрюшинных гематом и кровоизлияний в ЦНС. Пациенты страдают прогрессирующей артропатией и социально дезадаптированы. Им назначают неочищенные, не прошедшие вирусную инактивацию препараты: фактор свёртывания крови VIII (криопреципитат*), СЗП, концентрат нативной плазмы (КНП).
Выбор метода лечения зависит от формы и тяжести гемофилии, а также от локализации кровоизлияния или кровотечения.
Для лечения лёгкой формы гемофилии с уровнем фактора более 10% и женщин-носительниц гемофилии А с уровнем фактора VIII менее 50% применяют десмопрессин, который обеспечивает освобождение фактора VIII и фактора Виллебранда из депо эндотелиальных клеток. Десмопрессин вводят внутривенно капельно в дозе 0,3 мкг/кг в 50 мл изотонического раствора натрия хлорида за 15-30 мин. Десмопрессин показан при необширных хирургических вмешательствах и при проведении операций у женщин-носительниц. При тяжёлой форме гемофилии требуется лечение концентратами факторов VIII/IX.
Известно, что 1 ME фактора, введённая из расчета на 1 кг массы пациента, повышает активность фактора VIII в плазме крови на 2% гфи гемофилии А и фактора IX на 1% при гемофилии В. Дозу факторов VIII/IX определяют по формулам:
Доза ребёнку до года = масса тела х желаемый уровень фактора (%);
Доза ребёнку после года = масса телахжелаемый уровень фактора (%)х0,5.
Рекомендуемые дозы факторов VIII/IX в конкретных случаях разные. Все КФС вводят внутривенно струйно.
В начальной стадии острого гемартроза КФС вводят из расчёта 10 МЕ/кг, в поздней стадии — 20 МЕ/кг с повторным введением каждые 12 ч. Желаемый уровень фактора — 30-40%.
Пункция сустава
Показания к пункции сустава: первичный гемартроз; болевой синдром вследствие массивного гемартроза; рецидивирующий гемартроз; обострение хронического синовита.
В полость сустава после аспирации крови вводят гидрокортизон (гидрокортизона гемисукцинат) 50-100 мг через день, а для пролонгированного лечения — бетаметазон (дипроспан).
При наличии признаков хронического синовита в стадии обострения и рецидивирующих гемартрозах рекомендуют серию пункций 1-3 раза в неделю до полного купирования воспаления на фоне ежедневного гемостатического лечения (всего 4-6 пункций).
При отсутствии достаточного эффекта или невозможности адекватного лечения показана синовэктомия (радиоизотопная, артроскопическая или открытая). Через 1-2 дня после операции пациентам назначают физиотерапию и курс профилактического гемостатического лечения в течение 3-6 меС.
Лечение при кровоизлияниях в подвздошно-поясничную мышцу
КФС вводят в дозе 30-40 МЕ/кг каждые 8-12 ч в течение 2-3 сут при условии постельного режима и ограничения физических нагрузок.
Лечение при носовых кровотечениях
При носовых кровотечениях КФС вводят из расчёта 10-20 МЕ/кг каждые 8-12 ч с одновременным орошением слизистой оболочки носа карбазохромом (адроксоном), трансамином, этамзилатом (дициноном), 5% аминокапроновой кислотой и тромбином.
Лечение при кровотечениях слизистых оболочек полости рта
Такие кровотечения носят длительный характер. На месте повреждения часто образуется рыхлый сгусток, который не позволяет краям раны соединиться. После введения КФС, из расчёта 20-40 МЕ/кг каждые 8-12 ч, следует удалить сгусток и обеспечить соединение краёв раны. Антифибринолитические средства: аминокапроновая кислота, трансамин®. Фибриновый клей и охлаждённая протёртая пища способствуют местному гемостазу.
Санация ротовой полости
Перед лечением кариозного зуба достаточно однократного внутривенного введения концентратов факторов или, при гемофилии А, фактора свёртывания крови VIII (криопреципитата). В течение 72-96 ч до и после процедуры назначают аминокапроновую кислоту: детям 5% аминокапроновую кислоту внутривенно капельно в дозе 100 мг/кг на одну инфузию, взрослым — внутрь до 4-6 г/сут аминокапроновой кислоты за 4 приёма. Гемостатическое лечение начинают перед операцией и продолжают 2-3 сут после неё. Препарат вводят из расчета 10-15 МЕ/кг для экстракции резцов и 20 МЕ/кг при удалении больших коренных. Дополнительно используют местные и системные антифибринолитические средства, фибриновый клей. Рекомендуют строгую щадящую диету и холодное питьё.

Рис. 18-2. Протокол ведения больных гемофилией с почечным кровотечением (схема).
Лечение при почечных кровотечениях
Протокол лечения почечных кровотечений представлен на рис. 18-2. Гемостатическое воздействие проводят до купирования макрогематурии в дозе 40 МЕ/кг на введение. Желаемый уровень фактора — 40%. Кроме этого, назначают короткий курс преднизолона внутрь в дозе 1 мг/кг в сутки с последующей быстрой отменой.
Применение аминокапроновой кислотой пациентами с почечными кровотечениями противопоказано в связи с риском тромбоза почечных клубочков.
Желудочно-кишечные кровотечения
При желудочно-кишечных кровотечениях показано эндоскопическое исследование для уточнения причины и источника кровотечения. Протокол лечения представлен на рис. 18-3. Желаемый уровень фактора — 60-80%. Необходимо активное применение ингибиторов фибринолиза, а также лечение, общепринятое при эрозивно-язвенных заболеваниях желудка и кишечника.
Опасные для жизни кровотечения, в том числе кровоизлияния в головной мозг, и обширные хирургические вмешательства требуют введения КФС из расчёта 50-100 МЕ/кг 1-2 раза в сутки до купирования признаков кровотечения и последующего поддерживающего лечения меньшими дозами до заживления раны. Протокол ведения больного гемофилией с внутричерепным кровоизлиянием представлен на рис. 18-4. Непрерывная инфузия КФС в дозе 2 МЕ/кг в час обеспечивает их постоянный уровень не ниже 50% нормы. Кроме того, показано применение ингибиторов фибринолиза. В дальнейшем гемостатическое воздействие ведут по схеме профилактического лечения в течение 6 мес.
При отсутствии КФС используют фактор свёртывания крови VIII (криопреципитат4), СЗП и КНП (содержат фактор IX).
Средняя активность 1 дозы фактора свёртывания крови VIII (криопреципитата*) составляет 75 ME. Препарат поддерживает уровень фактора VIII в пределах 20-40%, что достаточно для проведения хирургических вмешательств. Вводят медленно, внутривенно струйно, в дозе 30-40 ед/кг через 8,12, 24 ч, в зависимости от желаемого уровня и вида кровотечения. 1 ед/кг препарата повышает уровень фактора на 1%.
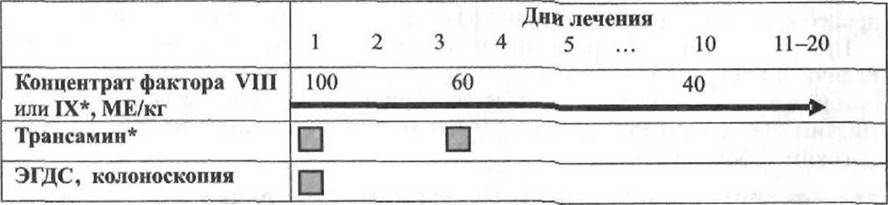
* Трансамин внутривенно капелыю в изотоническом растворе хлорида натрия: от 0-1 года — 0,15-0,4 г; 2-3 года — 0,3-0,7 г; 4-6 лет — 0,5-1,3 г; 7-14 лет — 0.8-2.0 г.
Рис. 18-3. Протокол ведения больных гемофилией с желудочно-кишечными кровотечениями (схема).
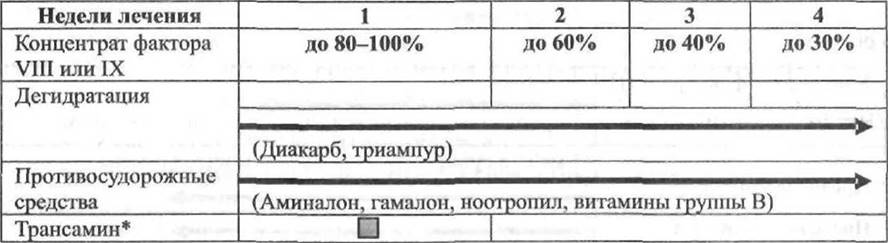
* Расчёт дозы трансамина см. на рис. 18-3.
Рис. 18-4. Протокол ведения больного гемофилией с внутричерепным кровоизлиянием.
При невозможности лечения больных гемофилией В концентратом фактора IX используют КНП из расчета 20-30 мл/кг в сутки в 2 приёма до стабилизации состояния, а при его отсутствии — СЗП. В1 дозе СЗП/КНП в среднем содержится 50-100 ME фактора IX. Вводят СЗП/КНП из расчёта 1 доза на 10 кг массы пациента.
Объективные ограничения применения фактора свёртывания крови VIII (криопреципитата*), СЗП и КНП:
• нестандартизованность и незначительный гемостатический эффект (приводят с детства к артропатии с ограничением движений, а в дальнейшем — к ранней инвалидности);
• низкая очистка препаратов и отсутствие противовирусной инактивации (поэтому 50-60% больных гемофилией имеют положительные маркёры гепатита С, 7% —постоянные носители вируса гепатита В;
• высокая частота аллергических и посттрансфузионных реакций;
• угроза перегрузки кровообращения из-за введения больших объёмов этих средств при минимальной концентрации в них факторов свёртывания;
• иммуносупрессия;
• низкое качество жизни пациентов.
Ингибиторная форма гемофилии
Появление у пациентов с гемофилией циркулирующих антикоагулянтов- ингибиторов, относящихся к классу имммуноглобулинов G, осложняют течение болезни. Частота ингибиторной формы гемофилии колеблется от 7 до 12%, а при очень тяжёлой гемофилии — до 35%. Ингибитор чаще появляется у детей 7-10 лет, но может быть обнаружен в любом возрасте. Появление ингибитора к факторам VIII/IX отягощает прогноз заболевания: кровотечения становятся профузными, сочетанными, развивается тяжёлая артропатия, приводящая к ранней инвалидности. Длительность циркуляции ингибитора составляет от нескольких месяцев до нескольких лет. Определение возможного ингибитора проводят в обязательном порядке у каждого пациента, как перед началом лечения, так и в его процессе, особенно при отсутствии эффекта от заместительного воздействия.
Присутствие ингибитора в крови подтверждают тестом Бетезда (Bethesda). Величина измерений — единица Бетезда (BE). Чем больше концентрация ингибитора в крови, тем больше количество единиц Бетезда (или выше титр Бетезда). Низким считается титр ингибитора менее 10 ед/мл, средним — от 10 до 50 ед/мл, высоким — более 50 ед/мл.
ЛЕЧЕНИЕ БОЛЬНЫХ ИНГИБИТОРНЫМИ ФОРМАМИ ГЕМОФИЛИИ
Повышение эффективности лечения пациентов с низким титром ингибитора достигают большими дозами концентратов фактора. Дозу подбирают эмпирически, чтобы полностью нейтрализовать ингибитор, а затем поддерживают концентрацию фактора VIII в крови пациента на заданном уровне в течение требуемого срока.
При кровотечениях, угрожающих жизни, или при необходимости хирургического вмешательства, обычно используют свиной фактор свёртывания крови VIII (Hyate-С), активированные препараты протромбинового комплекса: антиинги- биторный коагулянтный комплекс (Фейба Тим 4 Иммуно) и (Autoplex), эптаког альфа [активированный] (НовоСэвен). Свиной фактор свёртывания крови применяют при высоком тигре ингибиторов в крови (от 10 до 50 BE и выше). У 40% пациентов через 1-2 нед лечения появляется ингибитор к свиному фактору VIII. Лечение начинают с дозы 100 МЕ/кг (2-3 раза в сутки в течение 5-7 дней), которую при необходимости увеличивают. Для профилактики аллергических реакций и тромбоцитопении обязательна премедикация гидрокортизоном.
Концентраты протромбинового комплекса (КПК) и активированные концентраты протромбинового комплекса (аКПК) обеспечивают гемостаз в обход действия фактора VIII/IX. В их состав входят факторы VII и X в активированном виде, что значительно повышает эффективность лечения. Антиингибиторный коагулянтный комплекс (Фейба Тим 4 Иммуно4) вводят в дозе 40-50 МЕ/кг (максимальная разовая доза — 100 ед/кг) каждые 8-12 ч.
Эптаког альфа [активированный] (НовоСэвен) образует комплекс с тканевым фактором и активирует факторы IX или X. Препарат вводят каждые 2 ч. Дозы от 50 мкг/кг (при титре ингибитора менее 10 ВЕ/мл) и 100 мкг/кг (при титре 10-50 ВЕ/мл) до 200 мкг/кг (при титре более 100 ВЕ/мл). В комбинации с ним, как и с антиингибиторным коагулянтным комплексом (Фейба Тим 4 Иммуно), назначают антифибринолитические средства. Превышение доз антиингибиторного коагулянтного комплекса (Фейба Тим 4 Иммуно) и эптакога альфа [активированного] (НовоСэвен) приводит к тромботическим побочным реакциям.
В комплексном лечении ингибиторных форм гемофилии возможно применение плазмафереза. После удаления ингибитора пациенту вводят 10 000-15 000 ME концентрата фактора свёртывания крови VIII. Применяют различные виды иммуносупрессии: ГК, иммунодепрессанты.
Выработка иммунологической толерантности
По боннскому протоколу
В первый период вводят фактор свёртывания крови VIII по 100 МЕ/кг и антиингибиторный коагулянтный комплекс (Фейба Тим 4 Иммуно) по 40-60 МЕ/кг 2 раза в сутки ежедневно до снижения ингибитора до 1 ВЕ/мл.
Во второй период фактор свёртывания крови VIII вводят по 150 МЕ/кг 2 раза в сутки до полного исчезновения ингибитора. В дальнейшем большинство пациентов возвращают к профилактическому лечению.
Высокодозное лечение по протоколу Мальмё
Лечение назначают пациентам с титром ингибитора более 10 ВЕ/кг. Проводят экстракорпоральную абсорбцию антител с одновременным введением циклофосфамида (циклофосфана) (12-15 мг/кг внутривенно в первые двое суток, а затем 2-3 мг/кг применяют внутрь с 3 по 10-е сутки). Первоначальную дозу фактора свёртывания крови VIII рассчитывают так, чтобы полностью нейтрализовать оставшийся в циркуляции ингибитор и повысить уровень фактора свёртывания крови VIII более чем на 40%. Затем фактор свёртывания крови VIII вводят повторно, 2-3 раза в день, чтобы его уровень в крови сохранялся в пределах 30-80%. Кроме этого, сразу после первого применения фактора свёртывания крови VIII, пациенту внутривенно вводят нормальный человеческий иммуноглобулин G в дозе 2,5-5 г в первые сутки или 0,4 г/кг в сутки в течение 5 дней.
Лечение с использованием промежуточных доз препаратов фактора VIII включает их ежедневное введение в дозе 50 МЕ/кг.
Применение низких доз фактора свёртывания крови VIII предполагает первичное введении его в высокой дозе для нейтрализации ингибитора. В дальнейшем фактор вводят по 25 МЕ/кг каждые 12 ч на протяжении 1-2 нед ежедневно, далее - через день. Протокол применяют при угрожающих жизни кровотечениях и при хирургических вмешательствах.
Осложнения заместительного лечения
Появление ингибитора к дефицитным факторам в крови, развитие тромбоци- топении, гемолитическая анемия и вторичный ревматоидный синдром. Также к осложнениям относят инфицирование вирусами гепатитов В и С. ВИЧ, парвавиру- сом В 19 и цитомегаловирусом.
Практические рекомендации
• Больные и члены их семей должны получать специализированную медицинскую помощь в центрах по лечению гемофилии, где их обучают навыкам внутривенных инъекций и основам заместительного лечения.
• Воспитание детей обычное, отличается только тем, что с раннего детства необходимо избегать травм (обкладывать кроватку подушками, не давать игрушки с острыми углами и т.д.).
• Возможны занятия только неконтактными видами спорта, например, плаванием.
• Регулярно проводят профилактическую санацию зубов.
• Больных вакцинируют против вирусных гепатитов В и А.
• Оперативные вмешательства, экстракции зубов, профилактические прививки и любые внутримышечные инъекции проводят только после адекватного заместительного лечения.
• НПВП можно применять по строгим показаниям, только при наличии гемофилической артропатии и обострении хронического синовита. Необходимо избегать приёма дезагрегантов и антикоагулянтов.
• Перед оперативными вмешательствами и при неэффективности лечения проводят скрининг пациентов на наличие ингибитора к VIII или IX факторам.
• Два раза в год у больных определяют маркёры вирусных гепатитов В и С и ВИЧ а также делают биохимический анализ крови с исследованием печёночных проб.
• Раз в год больные проходят диспансеризацию.
• Оформляют инвалидность детства.
Ориентировочная потребность одного больного гемофилией в дефицитном факторе свёртывания крови в год считается равной 30 000 ME. Возможны также расчёты потребности в антигемофильных препаратах в зависимости от численности населения: 2 ME фактора на каждого жителя в год или 8500 доз фактора свёртывания крови VIII (криопреципитата*) на 1 млн жителей в год.
БОЛЕЗНЬ ВИЛЛЕБРАНДА
БВ — геморрагическое заболевание вследствие качественных или количественных нарушений фактора Виллебранда (ФВ). БВ бывает наследственная или приобретённая.
Наследственная болезнь Виллебранда
Причина наследственной БВ — полиморфизм гена, кодирующего синтез ФВ. Наследственная БВ является наиболее распространённым геморрагическим заболеванием. Частота носительства дефектного гена ФВ в популяции достигает 1 на 100 человек, но лишь 10-30% из них имеют клинические проявления.
ФВ экспрессируется в эндотелиоцитах и мегакариоцитах. Он содержится в альфа-гранулах тромбоцитов, эндотелиоцитах, в плазме и субъэндотелиальном матриксе. ФВ состоит из полимеров прогрессивно увеличивающейся молекулярной массы. Разделяют лёгкие, средние, тяжёлые и сверхтяжёлые мультимеры с молекулярной массой приблизительно от 540 кДа у димеров до нескольких тысяч килодальтон у самых крупных мультимеров. Чем больше молекулярная масса ФВ, тем больше их тромбогенный потенциал.
В гемостазе ФВ играет двоякую роль: опосредует адгезию тромбоцитов к субэндотелиальным структурам и взаимную адгезию тромбоцитов в процессе образования тромба, служит «носителем» фактора VIII в плазме, значительно удлиняя время его циркуляции.
КЛАССИФИКАЦИЯ И ПАТОГЕНЕЗ
Выделяют три типа БВ:
• Тип 1 — характеризуется количественным уменьшением содержания ФВ в крови различной степени тяжести;
• Тип 2 — характеризуется качественными изменениями ФВ. Вычленяют четыре подтипа: 2А, 2В, 2М, 2N;
• Тип 3 — практически полное отсутствие ФВ в крови.
Псевдоболезнь Виллебранда (тромбоцитарный тип) возникает вследствие повышенного связывания ФВ с гликопротеидом Ib-IX-V, что связано с изменениями структуры последнего. Это приводит к ускоренной элиминации, в первую очередь, наиболее высокомолекулярных комплексов ФВ из плазмы и диспропорциональному снижению его активности по сравнению с антигеном. При заболевании возможна умеренная тромбоцитопения. Псевдоболезнь Виллебранда фенотипически похожа на тип 2В БВ, но отличается от него локализацией нарушения. Для дифференциального диагноза необходимо провести RIPA с низкими концентрациями ристомицина. В этом тесте с плазмой здорового донора и тромбоцитами пациента агрегация будет наблюдаться у больного псевдо-болезнью Виллебранда, а при исследовании с тромбоцитами здорового донора и плазмой больного агрегация будет наблюдаться у пациента с БВ (тип 2В).
КЛИНИЧЕСКАЯ КАРТИНА
Основное клиническое проявление БВ — повышенная кровоточивость при травмах или патологических процессах. Поскольку при БВ страдает функция остановки кровотечения, для этого заболевания характерны первичные кровотечения, начинающиеся сразу после травмы.
Характер и тяжесть геморрагического синдрома при БВ зависят от формы заболевания. В целом, можно условно выделить три варианта.
• Геморрагический синдром по микроциркуляторному типу. Характерен для 1, 2А, 2В, 2М типов БВ. Типичны кожный гемисиндром в виде экхимозов, петехий, кровотечения из травмированных слизистых оболочек, длительные кровотечения из лунок удалённых или выпавших зубов, носовые кровотечения, маточные кровотечения у девочек после начала менструаций, интраоперационные и послеоперационные кровотечения, желудочно-кишечные кровотечения и кровотечения из мочевых путей. Менее характерны кровотечения из мест инъекций и гематомы мягких тканей после различных травм.
• Клинически напоминает гемофилию. У больных выражен геморрагический синдром по смешанному (гематомному и микроциркуляторному) типу. Характерен для пациентов с БВ 3-го типа, реже — для больных тяжёлыми формами других типов. Первые проявления заболевания возникают уже в периоде новорождённости: кожный гемисиндром, гематомы и кровотечения из мест инъекций. Позже — гематомы мягких тканей; кровотечения при травмах слизистых оболочек рта, при смене зубов; кровотечения из ран кожи и слизистых оболочек, кровотечения носовые, кишечные и из мочевых
путей. После начала менструаций у девочек нередки маточные кровотечения. Кровоизлияния в суставы, так же как и при гемофилии, могут появиться на первом году жизни. Для них характерны интраоперационные и послеоперационные кровотечения.
• Клиническая картина схожа с наблюдаемой у больных гемофилией А с сопоставимым уровнем фактора свёртывания крови VIII: гематомный тип кровоточивости, редко сопровождаемый поражением суставов. Характерен кожный гемсиндром в виде гематом, отсроченный, возникающей через несколько часов или дней после наступления травмы, кровотечения при травмах и после операций. У пациентов с БВ типа 2М могут возникать посттравматические гематомы мягких тканей.
Осложнения геморрагических проявлений
У детей с БВ возможно развитие хронической постгеморрагической анемии. Артропатии возникают у детей с рецидивирующими гемартрозами при 3-ем типе БВ. Описаны единичные случаи формирования псевдоопухолей.
Помимо геморрагических проявлений, нередко БВ сочетается с явлениями мезенхимальной дисплазии, что может влиять на выраженность геморрагического синдрома.
ДИАГНОСТИКА
Диагностические критерии Б В:
• типичный геморрагический синдром;
• снижение специфической активности ФВ (снижение vWF:RCo, vWF:CBA, vWF:FVIIIB);
• для типа 2В — положительная RIPA с низкими концентрациями ристоцетина.
Характеристика основных показателей системы гемостаза при его нарушениях вследствие различных причин представлена в табл. 18-3.
Активность ФВ связана с АВО групповой принадлежностью крови. У лиц с группой крови 1(0) конституционально снижено содержание ФВ.
Таблица 18-3. Нормы vWF:Ag, рекомендованные Всемирной ассоциацией тромбоза и гемостаза в зависимости от группы крови
|
Группа крови |
Нормальное содержание vWF:Ag |
|
0 |
36-157% |
|
А |
49-234% |
|
В |
57-241% |
|
АВ |
64-238% |
ЛЕЧЕНИЕ
Как любое наследственное заболевание, вылечить БВ нельзя; возможны лишь лечение или профилактика проявлений заболевания.
Специфическое гемостатическое воздействие (концентраты фактора Виллебранда, десмопрессин).
Препараты ФВ показаны при типе 3 и тяжёлом течении других типов БВ.
Введение СЗП при БВ с гемостатической целью не рационально из-за относительно низкой концентрации в ней ФВ. Фактор свёртывания крови VIII (криопреципитат*) содержит в 10 раз больше ФВ в единице объёма. Его недостатки — высокий риск инфицирования гемотрансфузионными инфекциями и содержание большого количества балластных веществ, в том числе с выраженной иммуногенной активностью. Поэтому, несмотря на низкую цену, его применение нерационально.
Наиболее эффективно использование очищенных вирус-инактивированных концентратов (фактор свёртывания крови VIII + фактор Виллебранда).
Десмопрессин
Синтетический аналог антидиуретического гормона вазопрессина — 1-дезамино-8-D-аргинин вазопрессин (десмопрессин) стимулирует высвобождение ФВ из депо, приводящее к повышению концентрации фактора в плазме крови. Наиболее эффективно применение десмопрессина при типе 1 БВ, но возможен эффект и при типе 2А. Препарат вводят 1 раз в сутки внутривенно капельно в дозе 0,3 мкг/кг в 50-100 мл изотонического раствора натрия хлорида в течение 20-30 мин, либо подкожно в такой же дозе без разведения. Существуют дозированные спреи, содержащие высококонцентрированный десмопрессин для интраназального введения в дозе 150-300 мкг. Длительное применение (в течение нескольких дней подряд) приводит к формированию тахифилаксии вследствие истощения ФВ в депо. Не рекомендуют использовать у детей до 3 лет.
Антифибринолитики
Аминокапроновая кислота вводят внутривенно капельно из расчёта 100 мг/кг в течение первого часа, затем 30 мг/кг в час. Максимальная суточная доза составляет 18 г. Можно принимать внутрь. Транексамовую кислоту можно принимать внутрь или внутривенно капельно в дозе 20-25 мг/кг каждые 8-12 ч. Показания к применению: маточные кровотечения, кровотечения из слизистых оболочек полости рта, носовые и желудочно-кишечные кровотечения. Транексамовую кислоту применяют, как правило, в сочетании со специфическим гемостатическим лечением, но в лёгких случаях — основным препаратом.
При кровотечениях из мочевых путей применение антифибринолитиков категорически противопоказано из-за риска обтурации сгустками крови мочевыводящих путей.
Местные гемостатические препараты
Местные гемостатические препараты, — фибриновый клей, аминометилбензойная кислота (гемостатическая губка с амбеном*) и другие, — показаны при оперативном лечении и в стоматологической практике. Этамзилат (дицинон*) применяют в качестве дополнительного гемостатического препарата при купировании кровотечений различной этиологии, часто эффективен для профилактики носовых кровотечений. Препарат вводят парентерально в дозе 3-5 мг/кг 3 раза в сутки. При энтеральном приёме дозу можно увеличить в 1,5-2 раза.
Осложнения лечения БВ
Введение ФВ с целью гемостаза у пациентов с 3-м типом БВ в 10-15% случаев вызывает образование ингибитора (блокирующих антител). При ингибиторе введение концентратов ФВ противопоказано из-за риска развития постинфузионных анафилактических реакций.
Для гемостаза возможно применение рекомбинантного активированного концентрата фактора VII (эптаког альфа активированный, НовоСэвен*) в средней дозе 90 мкг/кг каждые 2-4 ч до остановки кровотечения. Показано применение антифибринолитиков и воздействия, направленные на элиминацию ингибитора (применение гормонов, плазмаферез, внутривенное введение иммуноглобулина и др.).
Приобретённая болезнь Виллебранда
Приобретённая болезнь Виллебранда — геморрагическое состояние, лабораторно и клинически сходное с нарушениями, характерными для врождённой БВ. Всего описано около 300 случаев приобретённой БВ. У детей развитие приобретённой БВ происходит на фоне заболеваний сердца, сосудов, соединительной ткани, системных и онкологических процессов.
Патогенетические механизмы формирования дефицита ФВ:
• специфические антитела к фактору VIII/ФВ;
• неспецифические антитела, формирующие иммунные комплексы и активирующие клиренс ФВ;
• абсорбция ФВ клетками злокачественных опухолей;
• повышение протеолитической деградации ФВ;
• потеря тяжелых молекул ФВ при стрессе с высоким напряжением сдвига в условиях активного кровотока;
• снижение синтеза или высвобождения ФВ.
ДИАГНОСТИКА
Признаки приобретенной и наследственной БВ клинически сходны, и также не различаются методы лабораторной диагностики. Необходимо не только исследовать состояние гемостаза, но и диагностировать основное заболевание.
ЛЕЧЕНИЕ
Симптоматическое воздействие и/или профилактика кровотечений. В ряде случаев эффективно применение десмопрессина и концентрата фактора VIII + ФВ (фактор свёртывания крови VIII + фактор Виллебранда). Возможно применение антиингибиторного коагулянтного комплекса (Фейба Тим 4 Иммуно*) и эптакога [альфа активированного] (НовоСэвен*). Патогенетическое лечение включает воздействие на основное заболевание.
Таблица 18-4. Рекомендуемые дозы препаратов фактора Виллебранда для некоторых клинических ситуаций у детей
|
Характер кровотечения |
Доза, МЕ/кг |
Число введений |
Необходимый уровень в плазме крови |
|
большие операции, аденотонзиллото- мии (профилактика кровотечения) |
50-70 |
Раз в сутки |
>50% до наступления репарации |
|
Небольшие хирургические вмешательства (профилактика кровотечения) |
30-60 |
Раз в сутки |
>30-50% до наступления репарации |
|
Небольшие хирургические вмешательства (профилактика кровотечения) |
30-60 |
Раз в сутки |
>30-50% 2-3 дня |
|
Маточные кровотечения |
50-80 |
Раз в сутки |
>50% до прекращения |
|
Носовые кровотечения |
30-60 |
Однократно |
>30-50% |
ТРОМБОФИЛИЯ
Основная роль гемостаза заключается в сохранении жидкого состояния крови в сосудах и создания гемостатической «пробки», закрывающей дефект сосуда при травме или патологическом процессе, предотвращая кровопотерю. Гемостатическая пробка не должна мешать кровоснабжению органов.
Образование тромба — процесс динамический, в котором принимают участие три основных фактора: гемостатические компоненты крови, состояние сосудистой стенки и динамика тока крови (триада Вирхова). В норме компоненты находятся в динамическом равновесии, что способствует поддержанию гемостатического баланса. Нарушение любого из компонентов триады Вирхова может привести к изменению гемостатического баланса в сторону недостаточного или избыточного тромбообразования. В случае тромбофилии, как правило, происходит нарушение нескольких компонентов системы гемостаза, и часто нельзя вычленить ведущее нарушение.
Тромбофилия — хроническое состояние организма, при котором на протяжении длительного периода (месяцы, годы, в течение всей жизни) существует тенденция либо к спонтанному тромбообразованию, либо к неконтролируемому распространению тромба за пределы повреждения. Обычно под понятием «тромбофилия» понимают генетически детерминированное состояние, однако существуют приобретённые состояния повышенной склонности к тромбообразованию. Поэтому мы считаем, что рационально разделять тромбофилии на врождённые и приобретённые.
Нельзя ставить равенство между тромбофилией, тромбозом и тромбоэмболией, так как тромбофилия определяет лишь потенциальную возможность, которая не обязательно реализуется в виде тромбоза.
Тромбоз — патологическое состояние, связанное с нарушением кровотока и ишемией органа вследствие закрытия тромбом просвета сосуда. Тромбоэмболией называют обтурацию артериального сосуда тромбом, образовавшимся в лежащих выше отделах кровеносной системы и попавшим в сосуд с током крови.
Развитие тромбоза является следствием взаимодействия факторов патогенеза тромбообразования. Тромбозы бывают артериальные и венозные.
Артериальные и внутрисердечные тромбы состоят преимущественно из тромбоцитов, соединенных фибриновыми мостиками, — белые тромбы. Артериальные тромбы преимущественно пристеночные. Важнейшие факторы формирования артериального тромба — врождённая или приобретённая аномалия сосудистой стенки и патологическая активация тромбоцитов. Наиболее частая аномалия — атеросклероз. Кроме него возможны врождённые нарушения развития сосудов, ангиоматозные образования, инфекционное поражение эндотелия, ятрогенные нарушения.
Венозные тромбы включают значительное количество эритроцитов и фибрина; они часто полностью обтурируют просвет сосуда. Основной механизм образования венозного тромба связан с повышением свёртываемости крови и стазом. В детском возрасте ведущее значение приобретает катетеризация вен для проведения инфузий.
Тромбозы у детей встречаются гораздо реже, чем у взрослых. В первом полугодии жизни частота тромботических эпизодов составляет 5,1 на 100 000 детей в год, а после 6 мес колеблется от 0,7 до 1,9 на 100 000 детей в год. Венозные тромбозы у детей встречаются примерно в 2 раза чаще артериальных.
Факторы патогенеза патологического тромбообразования бывают врождённые и приобретённые. Среди врождённых факторов выделяют наследственные, как правило, связанные с генетически обусловленным изменением активности различных белков гемостаза или с повышением в крови концентрации веществ, обладающих протромботической активностью.
Факторы тромбофилии, связанные с изменением активности белков гемостаза, в свою очередь тоже можно подразделить на несколько групп:
• патологическое снижение активности антикоагулянтов;
• патологическое повышение активности прокоагулянтов;
• полиморфизм прокоагулянтов, защищающий их от воздействия ингибиторов.
Значение каждой группы факторов неодинаково: если роль факторов первой и второй категорий доказана, то факторы, второй категории, очевидно, менее значимы.
В эту группу факторов можно также отнести различные аномалии развития сосудов, существенно повышающие риск патологического тромбообразования, которые нельзя отнести к наследственным.
Приобрётенные факторы разнообразны. У детей они редко становятся единственной причиной патологического тромбообразования, но часто служат «последней каплей», ведущей к тромбозу или эмболии. Среди приобретённых факторов у детей ведущее место занимают внутривенные катетеры.
Наследственные факторы риска тромбообразования у детей:
• дефицит АТІІІ;
• дефицит протеина С;
• дефицит протенина S;
• полиморфизм гена фактора V (фактор V Лейдена);
• полиморфизм гена протромбина (однонуклеотидная замена G20210A);
• полиморфизм тромбоцитарного рецептора гликопротеина ІІІа;
• дисфибриногенемия;
• гиперлипопротеинемия;
• гипергомоцистеинемия (у детей, как правило, носит наследственный характер);
• талассемия (постспленэктомический тромбоз печёночных вен);
• серповидно-клеточная анемия.
Приобретённые факторы риска тромбообразования у детей:
• катетеризация вен, особенно длительное нахождение катетера в вене;
• повышение вязкости крови (полицитемия, потеря жидкости с уменьшением . ОЦК);
• операция или травма;
• инфекция (ВИЧ, ветряная оспа, гнойный тромбофлебит);
• аутоиммунные заболевания (волчаночный антикоагулянт, антифосфолипидный синдром, сахарный диабет, болезнь Бехчета и др.);
• нефротический синдром;
• врождённые пороки развития сердца и сосудов;
• онкологические заболевания;
• химиотерапия: аспарагиназа (L-аспарагиназа*), преднизолон;
• заболевания печени;
• назначение концентратов протеина С.
Факторы, роль которых в развитии тромбозов неясна:
• высокий уровень активности факторов свёртывания крови VIII, XI, XII, ФВ, ингибитора активатора плазминогена:
• дефицит факторов XII, кофактора гепарина II, плазминогена, активаторов плазминогена, тромбомодулина.
Важный фактор, учитываемый при риске патологического тромбообразования, возраст пациента. У детей риск тромбообразования наиболее велик в неонатальном периоде. Считается, что у новорождённых повышен риск тромбообразования вследствие низкой фибринолитической активности естественных антикоагулянтов (АТІІІ, протеины S и С (ПS, ПС) и относительно высокой активности факторов VIII и ФВ. Возможно, правильнее говорить о меньшей устойчивости гемостатического баланса, что связано с относительно низкой концентрацией многих белков гемостаза, ведущей к облегчению возникновения тромботических или геморрагических нарушений.
Возрастает риск развития тромботических осложнений у детей недоношенных или с задержкой внутриутробного развития.
Для возникновения тромбоза в детском возрасте необходимо взаимодействие целого ряда факторов. При изолированном факторе риска, как правило, тромбозы манифестируют во взрослом возрасте. Однако у пациентов с тяжёлым дефицитом АТIII, ПС, и ПS развитие спонтанных или спровоцированных минимальными воздействиями тромбозов возможно уже в раннем возрасте.
Среди приобретённых факторов риска тромбоза на первом месте у детей всех возрастов стоит катетеризация центральных вен. Этот фактор присутствует у 90% детей с тромбозами в возрасте до года и у 66% детей с тромбозами старше года. Более того, дети с обширными тромбозами вследствие катетеризации централь
ных вен имеют серьёзный риск длительно сохраняющихся осложнений, включая посттромботический синдром. В большинстве случаев тромбозы, связанные с установкой катетеров, возникают в системе верхней полой вены и в сердце. Система нижней полой вены может страдать при установке катетера в пупочную вену.
Лабораторная диагностика
Лабораторный анализ для выявления патогенетических факторов тромбоза необходимо проводить сразу же после диагностики, до начала лечения. Рекомендуемый набор тестов включает: АЧТВ, протромбинового времени, фибриноген, факторы свёртывания крови V, VII, VIII, IX, XI, XII, ФВ, исследование резистентности к активированному ПС, активность АТIII, ПС, ПS, плазминогена, д-димеры, время лизиса эуглобулинового сгустка, тесты для выявления волчаночного антикоагулянта — тест с ядом гадюки Рассела, тесты нейтрализации на фосфолипидах или тромбоцитах, исследование активности факторов при последовательных разведениях плазмы, микст-тесты для определения характера ингибитора. Определяют активность и наличие антигена активатора плазминогена и ингибитора активатора плазминогена-1. Необходимо определять уровень гомоцистеина в крови, а также генетический полиморфизм фактора V Лейдена, метилте- трагидрофолат редуктазы, протромбина (однонуклеотидная замена G20210A).
Лечение тромбозов у детей
В настоящее время проблема лечения детей недостаточно изучена. Возможно, что для детей старшего возраста допустимы подходы к лечению тромбозов, принятые у взрослых. Тем не менее, существуют данные, предполагающие различие реакций взрослых и детей (особенно до 6-месячного возраста) на антикоагулянтное и тромболитическое лечение. Возрастные особенности состояния системы гемостаза необходимо учитывать при назначении лечения.
Основная тактика ведения детей с тромбозами заключается в назначении на первом этапе гепаринотерапии с последующим переходом на длительное применение непрямых антикоагулянтов. Рекомендуют минимум 3 мес после прекращения действия факторов патогенеза тромбоза проводить поддерживающее лечение антикоагулянтами. При наличии нетяжёлых наследственных факторов тромбофилии воздействие антикоагулянтов должно быть продлено до 6 мес, а при сохраняющемся серьёзном риске рецидива тромбоза непрямые антикоагулянты можно применять годами.
Заместительное применение СЗП или концентратов протеина С (ПС), AT III можно проводить для лечения тромботических эпизодов, связанных с тяжёлым дефицитом ПС, ПS, AT III, для профилактики тромбозов при необходимости инвазивного лечения либо при присоединении дополнительных факторов риска тромбоза (например, инфекции), особенно у детей раннего возраста. У новорождённых и детей первых месяцев жизни антикоагулянтное и тромболитическое лечение может быть неэффективным из-за низкого возрастного уровня AT III и плазминогена. В этом случае показана инфузия СЗП.
В тромболитическом лечении артериальных и венозных тромбозов успешно применяют рекомбинантный тканевой активатор плазминогена (алтеплаза). Эффективно и относительно безопасно применение у детей сочетание проурокиназы и гепарина натрия (гепарина*).
Другими антикоагулянтами служат синтетические аналоги гирудина, блокирующие активные сайты тромбина, в том числе связанного с фибриногеном. Не влияют на АЧТВ и не связываются с тромбоцитами, редко вызывают геморрагические осложнения. Есть данные об их эффективном использовании у детей.
Анкрод - предотвращает образование перекрёстных связей фибрина и облегчает его расщепление плазмином. Хорошо зарекомендовал себя при гепаринин- дуцированной тромбоцитопении с тромбозом. Эффективность препарата у детей пока не исследована.
ЗАБОЛЕВАНИЯ ТРОМБОЦИТОВ
Тромбоциты, или кровяные пластинки, играют ключевую роль в процессах сосудисто-тромбоцитарного гемостаза — начальной стадии тромбообразования.
Тромбоциты представляют собой безъядерные клеточные элементы крови размером 1-4 мкм (молодые формы тромбоцитов крупнее) и продолжительностью жизни 7-10 сут. 1/3 тромбоцитов находится в селезёнке и 2/3 - в кровотоке. Количество тромбоцитов в периферической крови человека варьирует в пределах 150 000-400 000/мм3. Средний объём тромбоцита составляет 7,1 фемтолитров (1x1015 л).
При снижении количества тромбоцитов или нарушении их функции возможно возникновение кровотечений. Наиболее типичны кровотечения из повреждённой кожи и слизистых оболочек: петехии, пурпура, экхимозы, носовые, маточные, желудочно-кишечные кровотечения, гематурия. Внутричерепные кровоизлияния встречаются достаточно редко.
Иммунные тромбоцитопении
Наиболее частая причина развития тромбоцитопении — иммунная деструкция кровяных пластинок под действием ауто-, алло- или лекарственно-индуцированных антител. Возможный патогенез иммунной тромбоцитопении представлен на рис. 18-5. Ниже перечислены основные причины, приводящие к развитию тромбоцитопении.
Патофизиологическая классификация тромбоцитопенических состояний
I. Повышенное разрушение тромбоцитов (при нормальном или увеличенном количестве мегакариоцитов в костном мозге — мегакариоцитарная тромбоцитопения).
• Иммунные тромбоцитопении.
❖ Идиопатические:
-ИТП.
❖ Вторичные:
- индуцированы инфекциями, в том числе вирусными (ВИЧ, цитомегало- вирус [ЦМВ], Эпстайна-Барр, герпес, коревая краснуха, корь, эпидемический паротит, парвовирус В19) и бактериальными (туберкулёз, тиф);
- лекарственно-индуцированные;
- посттрансфузионная пурпура;
- аутоиммунная гемолитическая анемия (синдром Фишера-Эванса).
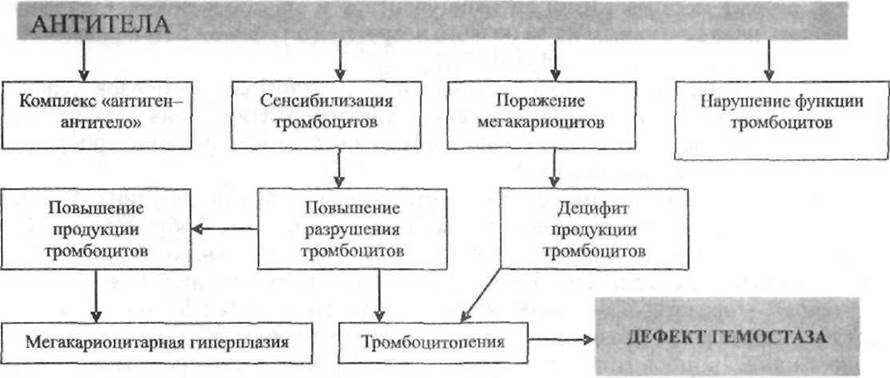
Рис. 18-5. Возможный патогенез иммунной тромбоцитопении.
-СКВ;
- гипертиреоз;
- лимфопролиферативные заболевания;
- аллергия, анафилаксия.
❖ Врождённые иммунные тромбоцитопении:
- врождённая трансиммунная тромбоцитопения;
- врождённая аллоиммунная тромбоцитопения;
- фетальный эритробластоз — резус-несовместимость.
• Неиммуные тромбоцитопении
❖ Вызванные повышенным потреблением тромбоцитов:
- микроангиопатическая гемолитическая анемия;
- ДВС-синдром;
- хроническая рецидивирующая шизоцитарная гемолитическая анемия;
- гемолитико-уремический синдром;
- ТТП;
- синдром Казабаха-Мерритта (гигантская гемангиома);
- «синие» пороки сердца.
❖ Вызванные повышенным разрушением тромбоцитов:
- лекарственные (ристоцетин, протамин сульфат, блеомицин и др.);
- стеноз аорты;
- инфекции;
- кардиальные (искусственные клапаны, репарация внутрисердечных дефектов и др.);
- злокачественная гипертония.
II. Нарушение распределения и депонирования тромбоцитов
• Гиперспленизм (портальная гипертензия, болезнь Гоше, врождённые «синие» пороки сердца, новообразования, инфекции и др.)
• Гипотермия.
III. Снижение продукции тромбоцитов (снижение или отсутствие мегакариоци- тов в костном мозге — амегакариоцитарная тромбоцитопения)
• Гйпоплазия или угнетение мегакариоцитов 1
❖ Лекарственные (хлортиазиды, эстрогены, этанол, толбутамид и др.).
❖ Конституциональные:
- тромбоцитопения при TAR-синдроме (врождённое отсутствие лучевых костей);
- врождённая гипоплазия тромбоцитов без аномалий;
- врождённая гипопластическая тромбоцитопения с микроцефалией;
- краснушная эмбриофетопатия;
- трисомия 13,18;
- анемия Фанкони.
❖ Неэффективный тромбоцитопоэз:
- мегалобластная анемия (дефицит витамина В12 и фолиевой кислоты);
- тяжёлая ЖДА;
- наследственные тромбоцитопении;
- пароксизмальная ночная гемоглобинурия.
❖ Нарушения регуляции тромбоцитопоэза:
- недостаточность тромбопоэтина;
- перемежающийся дисгенез тромбоцитов;
- циклическая тромбоцитопения.
❖ Метаболические нарушения:
- метилмалоновая ацидемия;
- кетоновая глицинемия;
- дефицит карбоксилсинтетазы;
- изовалериановая ацидемия;
- идиопатическая гиперглицинемия;
- новорождённые от матерей с гипотиреозом.
❖ Наследственные заболевания тромбоцитов 2:
- синдром Бернара-Сулье;
- аномалия Мея-Хегглина;
- синдром Вискотта-Олдрича;
- сцепленная с полом тромбоцитопения;
- средиземноморская тромбоцитопения.
❖ Приобретённые апластические заболевания:
- идиопатические;
- лекарственно-индуцированные (передозировка противоопухолевых препаратов, органический и неорганический мышьяк, мезантоин, триметин, антитиреоидные, противодиабетические, антигистаминные препараты, бутадион, инсектициды, препараты золота, идиосинкразия к левомице- тину);
- радиационные;
- вирус-индуцированные (гепатиты, ВИЧ, вирус Эпстайна-Барр и др.).
• Инфильтративные процессы в костном мозге
❖ Доброкачественные:
- остеопетроз;
- болезни накопления.
❖ Злокачественные:
- новообразования: лейкозы, миелофиброз, Лангергансово-клеточный гистиоцитоз.
- вторичные: лимфомы, нейробластома, метастазы солидных опухолей.
При снижении числа мегакариоцитов в костном мозге в дополнение к аспирации, для предотвращения типичных ошибок, обязательно выполняют биопсию костного мозга. Эти состояния ассоциированы с нормальным или повышенным количеством мегакариоцитов в костном мозге.
Идиопатическая (аутоиммунная) тромбоцитопеническая пурпура
ИТП — заболевание, характеризующееся изолированным снижением количества тромбоцитов (менее 100 000/мм3) при нормальном или повышенном количестве мегакариоцитов в костном мозге и наличием на поверхности тромбоцитов и в сыворотке крови антитромбоцитарных антител, вызывающих повышенную деструкцию тромбоцитов.
ИТП бывает острой, хронической и рецидивирующей. При острой форме количество тромбоцитов нормализуется (более 150 000/мм3) в течение 6 мес после постановки диагноза без возникновения рецидивов. При хронической форме тромбоцитопения менее 150 000/мм3 длится более 6 мес. При рецидивирующей форме количество тромбоцитов после возврата к нормальному уровню снова снижается. Для детей более характерна острая форма, для взрослых — хроническая. Клинические признаки острой и хронической форм ИТП представлены в табл. 18-6.
Вследствие того, что ИТП часто протекает транзиторно, истинная заболеваемость не установлена. Учтённая заболеваемость составляет около 1 на 10 000 случаев в год (3-4 на 10000 случаев в год среди детей до 15 лет).
Таблица 18-6. Клинические признаки острой и хронической идиопатической тромбоцитопенической пурпуры
|
Клинические признаки |
Острая ИТП |
Хроническая ИТП |
|
Возраст |
Дети 2-6 лет |
Взрослые |
|
Пол |
Роли не играет |
М/Ж- 1:3 |
|
Сезонность |
Весеннее время |
Роли не играет |
|
Предшествующие инфекции |
Около 80% |
Обычно нет |
|
Ассоциированные аутоиммунные состояния (СКВ и др.) |
Не характерно |
Характерно |
|
Начало |
Острое |
Постепенное |
|
Количество тромбоцитов, в мм3 |
Более 20 000 |
40 000-80 000 |
|
Эозинофилия и лимфоцитоз |
Характерно |
Редко |
|
Уровень IgA |
Нормальный |
Снижен |
|
Антитромбоцитарные антитела |
- |
- |
|
GpV |
Часто |
Нет |
|
Gpllb/llla |
Редко |
Часто |
|
Продолжительность |
Обычно 2-6 нед |
Месяцы и годы |
|
Прогноз |
Спонтанная ремиссия в 80% случаев |
Неустойчивое длительное течение |
Как сказано выше, в основе патогенеза ИТП — повышенное разрушение нагруженных аутоантителами тромбоцитов клетками ретикулогистиоцитарной системы. В опытах с меченными тромбоцитами установлено, что продолжительность жизни тромбоцитов снижается от 1-4 ч до нескольких минут. Повышение содержания иммуноглобулинов (IgG) на поверхности тромбоцитов и частота деструкции кровяных пластинок при ИТП пропорциональны уровню тромбоцит- ассоциированных IgG (PAIgG). Мишенями для аутоантител являются гликопротеины (Gp) мембраны тромбоцитов: Gp Hb/IIIa, Gp Ib/IX и Gp V.
Люди c HLA-фенотипом B8 и В12 имеют повышенный риск развития заболевания при наличии у них преципитирующих факторов (комплексы антиген-антитело).
Пик заболеваемости ИТП приходится на возраст от 2 до 8 лет, при этом мальчики и девочки болеют с одинаковой частотой. У детей младше 2 лет (инфантильная форма) заболевание характеризуется острым началом, тяжёлым клиническим течением с развитием глубокой тромбоцитопении менее 20 000/мм3, плохим ответом на воздействие и частой хронизацией процесса — до 30% случаев. Риск дебюта хронической ЙТП у детей также увеличен у девочек старше 10 лет при длительности заболевания более 2-4 нед до момента постановки диагноза и количестве тромбоцитов более 50 000/мм3. В табл. 18-7 показаны различия между хронической инфантильной и хронической детской ИТП.
В 50-80% случаев заболевание возникает через 2-3 нед после инфекционного заболевания или иммунизации (натуральная оспа, живая коревая вакцина и др.). Наиболее часто начало ИТП ассоциируют с неспецифическими инфекциями верхних дыхательных путей, приблизительно в 20% случаев — специфическими (коревая краснуха, корь, ветряная оспа, коклюш, эпидемический паротит, инфекционный мононуклеоз, бактериальные инфекции).
Таблица 18-7. Различия между хронической инфантильной и хронической детской идиопатической тромбоцитопенической пурпурой
|
Признаки |
Хроническая инфальтильная ИТП |
Хроническая детская ИТП |
|
Возраст (месяцы) |
4-24 |
Более 24 |
|
Мальчики/девочки |
3:1 |
3:1 |
|
Начало |
Внезапное |
Постепенное |
|
Предшествующие инфекции (вирусные) |
Обычно нет |
Часто |
|
Количество тромбоцитов при диагностике, 8 ММ3 |
Более 20 000 |
40 000-80 000 |
|
Ответ на лечение |
Плохой |
Временный |
|
Частота от общей заболеваемости, % |
30 |
10-15 |
КЛИНИЧЕСКАЯ КАРТИНА
Клинические проявления ИТП зависят от выраженности тромбоцитопении. Геморрагический синдром проявляется в виде множественных петехиально- синячковых высыпаний на коже, кровоизлияний на слизистых оболочках. Так как петехии (1-2 мм), пурпура (2-5 мм) и экхимозы (более 5 мм) могут также сопровождать другие геморрагические состояния, дифференциальный диагноз ставят по количеству тромбоцитов в периферической крови (рис. 18-6) и продолжительности кровотечения (рис. 18-7).
Кровоточивость появляется при снижении количества тромбоцитов менее 50 000/мм3. Угроза серьёзных кровотечений возникает при глубокой тромбоцитопении менее 30 000/мм3. В начале заболевания носовые, десневые, желудочно- кишечные и почечные кровотечения обычно нехарактерны, редко бывают рвота кофейной гущей и мелена.
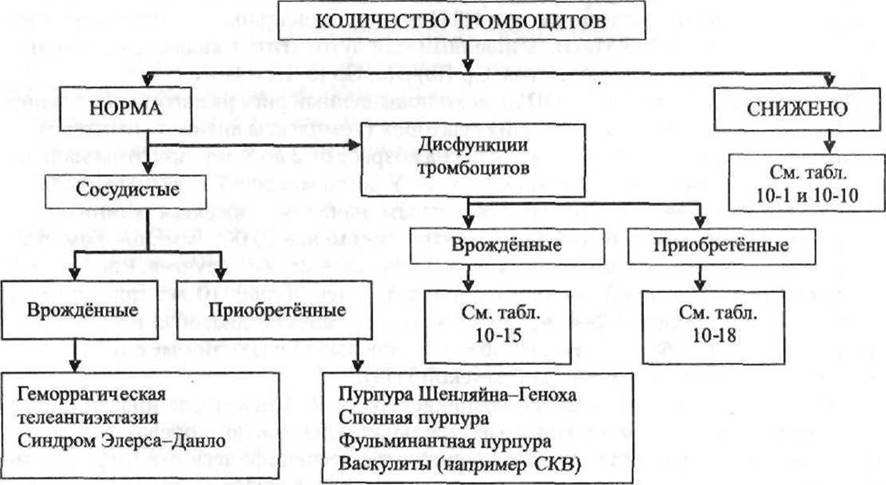
Рис. 18-6. Клинические подходы к дифференциальной диагностике геморрагического синдрома на основании количества тромбоцитов.

Рис. 18-7. Клинический подход к диагностике пурпуры на основании времени кровотечения.
Возможны тяжёлые маточные кровотечения. В 50% случаев заболевание проявляется в склонности к образованию экхимозов в местах ушибов, на передней поверхности нижних конечностей, над костными выступами. Глубокие мышечные гематомы и гемартрозы также не характерны, но могут быть следствием внутримышечных инъекций и обширных травм. При глубокой тромбоцитопении происходят кровоизлияния в сетчатку глаза, редко — кровотечение в среднее ухо, приводящее к снижению слуха. Кровоизлияние в мозг бывает в 1% случаев при острой ИТП, в 3-5% — при хронической ИТП. Обычно ему предшествует головная боль, головокружение и острое кровотечение какой-либо другой локализации.
При объективном обследовании у 10-12% детей, особенно раннего возраста, можно выявить спленомегалию. В этом случае дифференциальный диагноз проводят с лейкозом, инфекционным мононуклеозом, СКВ, синдромом гиперспленизма. Увеличение лимфатических узлов при ИТП быть не должно, если только это не связано с перенесённой вирусной инфекцией.
ЛАБОРАТОРНАЯ ДИАГНОСТИКА
При лабораторном обследовании выявляют тромбоцитопению менее 100 000/мм3, увеличение среднего объёма тромбоцита (MPV) по данным автоматического анализатора крови до 8,9±1,5 мкм3. В табл. 18-8 приведена классификация заболеваний тромбоцитов в зависимости от их размера.
Таблица 18-8. Классификация заболеваний тромбоцитов в зависимости от их размера
|
MPV повышен |
MPV норма (7,1 мкм) |
MPV снижен |
|
ИТП и другие состояния с повышенным производством и потреблением тромбоцитов, в том числе ДВС- синдром |
Состояния, при которых костный мозг малоклеточный или поражён злокачественными заболеваниями |
Синдром Вискотта-Олдрича |
|
Синдром Бернаре-Сулье |
- |
TAR-синдром |
Окончание табл. 18-8
|
Аномалия Мея-Хегглина |
- |
Болезни накопления |
|
Синдром Альпорта |
- |
Железодефицитная анемия |
|
Мукополисахаидозы, синдром «серых» тромбоцитов |
- |
- |
|
Синдром «дырявых» тромбоцитов («швейцарский» сыр) |
- |
- |
|
Синдром Монреальских тромбоцитов |
- |
- |
В периферической крови у пациентов с ИТП, помимо тромбоцитопении, может быть умеренная эозинофилия. При выраженной кровопотере развивается анемия.
В пунктате костного мозга, который проводят для исключения других онкогематологических заболеваний, находят раздражение мегакариоцитарного ростка, слабую «отшнуровку» тромбоцитов при нормальных эритроидном и миелоидном ростках. У части больных обнаруживают умеренную эозинофилию.
При исследовании коагуляционного профиля, необязательном при стандартном ИТП, выявляют увеличение времени кровотечения, снижение или отсутствие ретракции сгустка, нарушение утилизации протромбина при нормальных уровнях фибриногена, протромбинового времени и активированного парциального тромбопластинового времени.
Лабораторные исследования у пациентов с тромбоцитопенией включают:
• общий анализ крови с мазком и определением количества тромбоцитов;
• исследование пунктата костного мозга;
• анализ крови на АНФ, анти-ДНК, фракции комплемента С3, С4, антитромбоцитарные антитела, уровень плазменного гликокалицина, проведение пробы Кумбса;
• определение протромбинового времени, активированного парциального тромбопластинового времени, уровня фибриногена, продуктов распада фибриногена;
• определение мочевины, креатинина крови, печёночных проб;
• анализ крови на оппортунистические инфекции (ВИЧ, Эпстайна-Барр вирус, парвовирус);
• исключение вторичных форм тромбоцитопении.
Основные критерии для постановки диагноза ИТП:
• отсутствие клинических признаков системных и онкогематологических заболеваний;
• изолированная тромбоцитопения при нормальном количестве эритроцитов и лейкоцитов;
• нормальное или повышенное количество мегакариоцитов в костном мозге при нормальных эритроидных и миелоидных элементах;
• исключение вторичных форм тромбоцитопении при гиперспленизме, микро- ангиопатической гемолитической анемии, ДВС-синдроме, лекарственно- индуцированной тромбоцитопении, СКВ, вирусных инфекциях (Эпстайна- Барр вирус, ВИЧ, парвовирус).
ЛЕЧЕНИЕ
Поскольку в основе патогенеза ИТП лежит деструкция нагруженных аутоантителами тромбоцитов клетками ретикулогистиоцитарной системы, основными принципами лечения являются:
• уменьшение продукции аутоантител;
• нарушение связывания аутоантител с тромбоцитами;
• устранение деструкции сенсибилизированных антителами тромбоцитов клетками ретикулогистиоцитарной системы.
При отсутствии кровотечений из слизистых оболочек, слабо выраженных экхимозах после ушибов, количестве тромбоцитов более 35 000/мм3 лечение обычно не требуется. Больным следует избегать занятий контактными видами спорта. Менструирующим девочкам полезны длительно действующие препараты прогестерона (Депо-провера и другие) для задержки менструации на несколько месяцев с целью профилактики интенсивных маточных кровотечений.
Глюкокортикоиды
Механизм действия
• Угнетение фагоцитоза тромбоцитов с фиксированными на их поверхности антителами в селезёнке.
• Нарушение выработки антител.
• Нарушение связывания аутоантител с антигеном.
Показания
Кровотечения из слизистых оболочек; выраженная пурпура и обильные гематомы на местах ушибов, особенно на голове и шее; прогрессирующая пурпура; тромбоцитопения более 3 нед; рецидивирующая тромбоцитопения; количество тромбоцитов менее 20 000/мм3 у первичных пациентов с минимальной пурпурой.
Режимы введения
• Стандартные дозы пероральных ГК - преднизолон 1-2 мг/кг в сутки или 60 мг/м2 в сутки в течение 21 дня с постепенной отменой. Дозу снижают независимо от количества тромбоцитов, ремиссию оценивают по окончании курса. При отсутствии ремиссии или снижение количества тромбоцитов после достижения нормальных показателей глюкокортикоидное воздействие не продолжают. При отсутствии полного гематологического ответа во время стандартного курса ГК отмену преднизолона производят «прерывистым курсом» (через день после перерыва по 5 мг). Возможно повторение курса ГК спустя 4 нед. Длительное применение ГК при ИТП нежелательно, так как может привести к депрессии тромбоцитопоэза.
• Высокие дозы пероральных ГК 4-8 мг/кг в сутки в течение 7 дней или 10-30 мг/кг в сутки метилпреднизолона в течение 3-7 дней с быстрой отменой препарата. Через неделю курсы повторяют (2-3 курса).
• Высокие дозы парентеральных ГК 10-30 мг/кг в сутки метилпреднизолона или солюмедрол 500 мг/м2 в сутки внутривенно в течение 3-7 дней в тяжёлых случаях для более быстрого купирования геморрагического синдрома. При необходимости дальнейшего лечения пациента переводят на приём стандартных доз внутрь.
• Для стероидорезистентных пациентов с ИТП возможна «пульс-терапия» дексаметазоном — 6 циклов по 0,5 мг/кг в сутки (максимально 40 мг/сут) по 4 дня каждые 28 дней, приём внутрь.
Эффективность приёма ГК, по данным разных авторов, составляет 50-80%. Побочные эффекты при их применении: симптомы гиперкортицизма, язвенная болезнь, гипергликемия, гипертензия, увеличение риска инфекции, миопатия, гипокалиемия, стероидный психоз, нарушение функции яичников у девочек, задержка роста.
Внутривенный иммуноглобулин
Механизм действия;
• обратимая блокада Fc-рецепторов макрофагов;
• подавление синтеза аутоантител В-лимфоцитами;
• защита тромбоцитов и/или мегакариоцитов от антител;
• модуляция хелперной и супрессорной активности Т-лимфоцитов;
• подавление комплемент-зависимого повреждения тканей;
• выздоровление от персистирующих вирусных инфекций за счёт введения специфических антител.
Показания при острой ИТП:
• при возможности — воздействие первой линии;
• неонатальная симптоматическая иммунная тромбоцитопения;
• дети в возрасте до 2 лет. резистентные к воздействию ГК.
Современные препараты внутривенного иммуноглобулина (ВВИГ) должны соответствовать требованиям ВОЗ, определённым в 1982 г.: минимум 1000 порций крови, минимум 90% иммуноглобулинов G, нативный иммуноглобулин G (высокая активность Fc-фрагмента), нормальное деление иммуноглобулинов G на подклассы, физиологичный период полураспада. Кроме того, ВВИГ должны иметь низкую антикомплементарную активность и двойную вирус-инактивацию (чистый иммуноглобулин G). В табл. 18-9 представлены препараты ВВИГ, разрешённые к применению в России. Характеристика препаратов ВВИГ представлена в табл. 18-10.
Таблица 18-9. Препараты внутривенного иммуноглобулина, разрешённые к применению в России
|
Готовые к употреблению |
В виде концентратов |
|
Иммуноглобулин человека нормальный (интрагло- бин“) («Biotest», Германия), иммуноглобулин человека нормальный для внутривенного введения (имбио- гам*) («ИМБИО», Россия), (октагам*) («Octapharma», Швейцария), ИГ ВЕНА Н.И.В." («Kedrion», Италия) |
Иммуноглобулин («Biochemie», Австрия) сандоглобулин («Sandoz», Швейцария), иммуноглобулин человека нормальный (Эндобулин С/Д*) (Австрия), (Биавен B.H.*(«Pharma Biajini», Италия), (Веноглобулин*) («Paster Merieux», Франция), иммуноглобулин человека нормальный (Габриглобин*) («Ивановская ОСПК», Россия) |
Таблица 18-10. Сравнительная характеристика препаратов внутривенного иммуноглобулина
|
ИГ вена |
Иммуноглобулин человека нормальный (окта- гам*) |
Иммуноглобулин человека нормальный (интраглобин*) |
Сандоглобулин* |
|
|
IgG , мг/мл |
49-51 |
51-53 |
41-42 |
45-47 |
|
Fc интегрированные молекулы, % |
98-101 |
99-102 |
68-87 |
81-88 |
|
IgA, мг/мл |
0-0,015 |
0,05-0,1 |
1,5-2,0 |
0,5-0,75 |
|
IgM, мг/мл |
0 |
0,01-0,02 |
0,06-0,08 |
0,01-0,02 |
|
Стабилизатор |
Мальтоза |
Мальтоза |
Глюкоза |
Сукроза |
|
Титр CMV-антител, ЕД/мл |
50,0 |
22,0-23,0 |
12,0 |
Более 10,0 |
Режимы введения внутривенного иммуноглобулина
• При острой ИТП — общая доза 1-2 г/кг на курс по схеме: 400 мг/кг в сутки в течение 5 дней или 1 г/кг в сутки в течение 1-2 дней. Дети моложе 2 лет легче переносят 5-дневный протокол приёма препаратов I и II поколений.
• При хронической ИТП — начальная доза 1 г/кг в сутки в течение 1-2 дней, затем единичные инфузии в дозе 0,4-1 г/кг, в зависимости от ответа, для поддержания безопасного уровня тромбоцитов (более 30 000/мм3). Использование ВВИГ полезно сочетать с альтернирующими курсами ГК.
Ответ на воздействие у пациентов с острой ИТП происходит в 80-96,5% случаев. По сравнению с применением ГК быстрее увеличивается количество тромбоцитов при эпизодах кровотечения сопоставимой длительности. Около 65% детей с ИТП, резистентных к ГК, достигают длительной ремиссии после курса ВВИГ.
Побочные эффекты препаратов ВВИГ :
• анафилактические реакции (у пациентов со сниженным уровнем IgA);
• головная боль (20% случаев);
• лихорадка с ознобом (1-3% случаев);
• гемолитическая анемия с положительной пробой Кумбса.
В научной литературе был описан случай развития асептического менингита после инфузии ВВИГ, а также инфицирование реципиентов ВВИГ (Гаммагард, «Бакстер») вирусом гепатита С, но с 1994 г., после совершенствования технологии производства препаратов, такие ситуации больше не встречались.
Профилактическое назначение парацетамола (10-15 мг/кг каждые 4 ч) и дифенгидрамина (димедрола*) (1 мг/кг каждые 6-8 ч) уменьшает частоту и тяжесть лихорадки с ознобом, а внутривенное введение дексаметазона в дозе 0,15-0,3 мг/кг позволяет купировать головную боль при инфузиях ВВИГ.
Комбинированное применение глюкокортикоидов и внутривенного иммуноглобулина
Показания:
• кровотечение из слизистых оболочек;
• обширные петехии, пурпура и экхимозы;
• симптомы и/или признаки внутренних кровотечений, в особенности внутричерепных.
Комбинированное применение вызывает более быстрое увеличение числа тромбоцитов, чем каждый препарат по отдельности. Его применяют при угрожающих жизни кровотечениях и при подготовке к хирургическому вмешательству. В неотложных случаях в качестве глюкокортикоида можно применять метилпреднизолон 30 мг/кг в сутки в течение 3 дней или солюмедрол в дозе 500 мг/м2.
Анти-RhD-иммуноглобулин
Механизм действия:
• блокада Fc-рецепторов макрофагов нагруженными антителами эритроцитами;
• подавление образования антитромбоцитарных антител;
• иммуномодулирующий эффект.
Условия применения при ИТП — RhD-позитивные не спленэктомированные пациенты.
Препараты анти-RhD-иммуноглобулина: «WinRho» (Winnipeg, Manitoba, Канада), «NABI» (Boca Ration, FL, США), «Partogamma» (Biagini, Pisa, Италия), «Resogam» (Genteon Pharma, Германия). В России препараты анти-RhD- иммуноглобулина производят на станциях переливания крови (Ивановской, Московской и др.). 1500 ME препарата соответствуют 300 мкг анти-D- иммуноглобулина.
Режим введения:
• оптимальная курсовая доза 50 мкг/кг на курс в виде однократной внутривенной инфузии или дробного внутримышечного введения в течение 2-5 дней;
• при концентрации гемоглобина в крови больного менее 100 г/л, доза препарата составляет 25-40 мкг/кг на курс, при гемоглобине 100 г/л — 40-80- 100 мкг/курс;
• повторные курсы анти-D-иммуноглобулина с интервалом 3-8 нед для поддержания количества тромбоцитов более 30 000/мм3.
Количество тромбоцитов и уровень гемоглобина контролируют на 3-4-е сут после начала воздействия. Отсутствие гематологического ответа на первый курс анти-D-иммуноглобулина не является противопоказанием для проведения второго курса, так как 25% пациентов, не ответивших на лечение, достигают гематологического ответа при повторном введении препарата. Среди пациентов, резистентных к ГК, 64% достигают ремиссии после курса анти-D-иммуноглобулина. Значимое увеличение количества тромбоцитов отмечают через 48 ч после введения препарата, поэтому его не рекомендуют применять в угрожающих жизни ситуациях.
Побочные реакции:
• гриппоподобный синдром (температура, озноб, головная боль);
• падение уровня гемоглобина и гематокрита вследствие гемолиза, подтверждаемого положительной пробой Кумбса.
Случаев инфицирования вирусами при применении препаратов анти-D- иммуноглобулина не зарегистрировано. Острые аллергические реакции маловероятны. Описаны IgE-опосредованные и вызванные иммунными комплексами аллергические реакции. У пациентов с дефицитом IgA аллергические реакции не описаны. Гемолиз обычно внесосудистый. В описанных немногочисленных случаях внутрисосудистого гемолиза хроническая почечная недостаточность не развивалась. Среднее снижение уровня гемоглобина составляет 5-20 г/л и бывает кратковременным (1-2 нед).
Применение анти-RhD-иммуноглобулина безопасно, удобно, дёшево и эффективно у 79-90% пациентов с хронической ИТП, причём у детей больше, чем у взрослых.
Механизмы действия ГК, ВВИГ и анти-D-иммуноглобулина представлены в табл. 18-11.
Таблица 18-11. Механизм действия глюкокортикоидов, внутривенного иммуноглобулина и анти-D- иммуноглобулина
|
Эффект |
ГК |
Внутривенный иммуноглобулин |
Анти-D- иммуноглобулин |
|
Повышение резистентности капилляров |
+ |
- |
- |
|
Блокада ретикулоэндотелия |
+/- |
+ |
+ |
|
Связывание антител к тромбоцитам |
+ |
+/- |
- |
|
Нарушение связывания Fc R |
+ |
+ |
+/- |
|
Угнетение Т-лимфоцитов |
+ |
+ |
- |
|
Синтез иммуноглобулинов |
Повышается |
Повышается |
Норма/повышается |
|
Продукция цитокинов |
Повышается |
Повышается |
Норма |
Интерферон-альфа
Интерферон-альфа-2b может применяться при лечении пациентов с хронической ИТП, резистентных к ГК. Гематологический ответ достигается у 72% пациентов, в том числе у 33% не ответивших на ГК.
Механизм действия при ИТП: подавление продукции аутоантител за счёт ингибирующего эффекта интерферон-альфа-2Ь на выработку иммуноглобулинов В-лимфоцитами.
Режим введения: 0,5-2x106 ЕД, в зависимости от возраста, подкожно или внутримышечно 3 раза в неделю (обычно понедельник-среда-пятница) в течение
1-1,5 мес. Гематологический ответ отмечают на 7-39-й день от начала лечения. При отсутствии гематологического ответа лечение прекращают, при наличии — продолжают до 3 мес. После окончания курса препарат либо отменяют, либо назначают в поддерживающей дозе с уменьшением кратности введения до 1-2 раз в неделю (подбирают индивидуально). При рецидиве заболевания (обычно через
2-8 нед после окончания применения) показан повторный курс, который имеет такую же эффективность. Длительность поддерживающего лечения интерферон- альфа-2b при наличии гематологического ответа не определена.
Побочные эффекты: гриппоподобный синдром (лихорадка, озноб, головная боль, миалгии), боль и покраснение в месте инъекции, печёночная токсичность, угнетение миелопоэза (при дозах, превышающих 2x106 ЕД), депрессии у подростков.
Для уменьшения выраженности побочных эффектов (гриппоподобный синдром) перед первыми введениями препарата рекомендуют профилактическое назначение парацетамола.
Даназол
Даназол представляет собой синтетический андроген со слабой вирилизирующей активностью и иммуномодулирующим действием (восстановление функции Т-супрессоров).
Механизм действия даназола при идиопатической тромбоцитопенической пурпуре:
• модулирует экспрессию Fc-гамма-рецепторов на мононуклеарных фагоцитах и препятствует деструкции нагруженных антителами тромбоцитов;
• подавляет продукцию аутоантител;
• обладает синергизмом с ГК, способствует освобождению стероидов от связи с глобулинами и увеличивает их доступ к тканям.
Режим введения:
10-20 мг/кг в сутки внутрь (300-400 мг/м2) в 2-3 приёма в течение 3 мес и более для стабилизации эффекта.
Побочные эффекты:
акне, гирсутизм, увеличение веса, печёночная токсичность.
Гематологический ответ происходит примерно у половины детей с хронической ИТП, в том числе у пациентов, резистентных к ГК. Эффективность лечения увеличивается после проведения спленэктомии. В большинстве случаев ответ неполный.
Винкристин
Применяют винкристин в дозе 0,02 мг/кг (максимально 2 мг) внутривенно, еженедельно, всего 4 введения.
Винбластин
Винбластин применяют в дозе 0,1 мг/кг (максимально 10 мг) внутривенно, еженедельно, всего 4 введения.
В случае эффективности воздействия винкристина и винбластина происходит быстрое увеличение количества тромбоцитов, часто до нормального уровня. Большинство детей нуждаются в повторных введениях препарата с 2-3-недельным интервалом для поддержания безопасного количества тромбоцитов. При отсутствии ответа на лечение в течение 4 нед дальнейшее использование препаратов не показано.
Полная гематологическая ремиссия в течение 0,5-4 лет описана приблизительно у 10% пациентов, транзиторный ответ — у половины.
Побочные эффекты: периферическая нейропатия, лейкопения, алопеция, запоры, некрозы при попадании в подкожную клетчатку.
Циклофосфамид
Циклофосфамид (циклофосфан) применяют в качестве иммунодепрессанта. Гематологический ответ у пациентов с хронической ИТП во время лечения достигает 60-80% и сохраняется дольше по сравнению с другими препаратами. Полный гематологический ответ после окончания лечения происходит в 20-40% случаев. Лучшие результаты показаны у спленэктомированных пациентов с малой длительностью заболевания.
Механизм действия — подавление пролиферации лимфоцитарных клонов, участвующих в иммунном ответе.
Режим введения: 1-2 мк/кг в сутки, принимают внутрь. Гематологический ответ достигают через 2-10 нед от начала курса.
Побочные эффекты: угнетение миелопоэза, алопеция, печёночная токсичность, геморрагический цистит, лейкемия (отдалённое осложнение).
Азатиоприн
У пациентов с аутоиммунными заболеваниями азатиоприн применяют в качестве иммунодепрессанта. Увеличение числа тромбоцитов отмечают у 50% пациентов с ИТП, а полный гематологический ответ — у 10-20%.
Режим введения: 1-5 мг/кг в сутки (200-400 мг). До достижения максимального ответа продолжительность лечения может составлять 3-6 мес. Так как после окончания применения препарата заболевание рецидивирует, необходимо поддерживающее лечение.
Побочные эффекты: анорексия, тошнота, рвота, умеренная нейтропения, лимфомы (отдалённое осложнение).
Преимущество данного препарата у детей заключается в более низкой частоте развития опухолей по сравнению с циклофосфамидом (циклофосфаном*).
Циклоспорин
Циклоспорин (циклоспорин А) — нестероидный иммуносупрессант, вызывающий угнетение клеточного иммунитета. Препарат действует на активированные Т-лимфоциты-эффекторы, подавляя продукцию цитокинов (интерлейкина-2, интерферона-гамма, фактора некроза опухоли).
Режим введения: принимают внутрь в дозе 5 мг/кг в сутки в течение нескольких месяцев. Гематологический ответ наблюдается через 2-4 нед от начала приёма в виде некоторой стабилизации клинико-гематологических показателей, снижения уровня антитромбоцитарных антител. Рецидивы заболевания возникают сразу после отмены препарата.
Побочные эффекты: гипомагнийемия, гипертензия, печёночная и почечная токсичность, вторичные опухоли (отдалённые осложнения). Серьёзность побочных эффектов и неубедительный эффект, вызываемый применением циклоспорина, делает его применение при ИТП нежелательным.
Трансфузии тромбоцитов
Проведение трансфузии тромбоцитов показано в случае развития неврологической симптоматики, указывающей на возможность внутричерепных кровоизлияний, а также при проведении оперативных вмешательств у пациентов с глубокой тромбоцитопенией, резистентных к консервативному лечению. Хотя продолжительность жизни кровяных пластинок невелика, трансфузии тромбоцитов могут оказать временный гемостатический эффект. При этом боязнь увеличения продолжительности ИТП вследствие риска сенсибилизации только теоретическая. Трансфузии тромбоцитов применяют у пациентов с ИТП высокого риска с положительным клиническим эффектом. Переливание тромбоконцентрата осуществляют дробно по 1-2 дозы в час или по 6-8 доз каждые 4-6 ч до достижения клинико-гематологического ответа. Эффект трансфузии усиливают предварительным введением ВВИГ.
Спленэктомия
В случае отсутствия эффекта от консервативного лечения, наличия глубокой тромбоцитопении, геморрагического синдрома и угрозы развития опасных для жизни кровотечений, больным показано проведение спленэктомии. Вопрос об операции решают индивидуально при каждом случае.
Показания к спленэктомии:
• тяжёлая острая ИТП с наличием жизнеугрожающих кровотечений при отсутствии ответа на медикаментозное воздействие;
• длительность заболевания больше 12 мес, тромбоцитопения менее 10 000/мм3 и кровотечения в анамнезе;
• хроническая ИТП с признаками кровоточивости и постоянным уровнем тромбоцитов менее 30 000/мм3 при отсутствии ответа на лечение в течение нескольких лет.
У ведущих активный образ жизни, часто травмирующихся пациентов спленэктомия может быть произведена ранее.
Вследствие риска развития после операции генерализованных инфекций, спленэктомию проводят только при наличии чётких показаний. Операция редко бывает необходима в течение 2 лет с момента постановки диагноза, так как тромбоцитопения хорошо переносится и легко контролируется благодаря применению ГК и ВВИГ. Спонтанное восстановление количества тромбоцитов может наступить через 4-5 лет, следовательно, необходим очень осторожный подход к выполнению операции. У детей с хронической ИТП случаи спонтанной ремиссии отмечают в 10-30% случаев через несколько месяцев или лет после постановки диагноза, у взрослых очень редко.
Подготовка к спленэктомии включает в себя назначение ГК, ВВИГ или анти- D-иммуноглобулина. ГК назначают в полной дозе за день до, в день операции и в течение нескольких дней после её проведения, так как большинство пациентов имеют надпочечниковую недостаточность вследствие предыдущего их применения. При появлении активного кровотечения непосредственно перед операцией может потребоваться трансфузия тромбоцитов и эритромассы, а также введение метилпреднизолона (солюмедрола) в дозе 500 мг/м2 в сутки. Перед плановой операцией обязательно ультразвуковое исследование органов брюшной полости для выявления дополнительных селезёнок (15% случаев), а в спорных случаях — радиоизотопное скенирование.
Полное и длительное восстановление количества тромбоцитов после спленэктомии бывает приблизительно у 50% пациентов. Хорошим прогностическим признаком считается ответ на приём ГК и ВВИГ до операции (эффективность спленэктомии в 80-90%), а также отсутствие антитромбоцитарных антител после её проведения. 25% детей, перенёсших спленэктомию, не достигают клиникогематологического ответа и нуждаются в дальнейшем лечении.
Предпочтительно выполнение операции лапароскопическим методом (возможно у 90% пациентов) позволяет уменьшить объём оперативного вмешательства, уровень операционной кровопотери, обеспечить пациенту более быстрое возвращение к активной жизни и сократить сроки госпитализации. Послеоперационный рубец при этом имеет длину около 1 см и не вызывает дискомфорта.
Случаи смертельного исхода от бактериальных инфекций в позднем послеоперационном периоде, особенно у детей, перенесших спленэктомию до 5 лет, составляет 1:300 больных в год. Большинство из них происходит в течение 2 лет после операции. К основным причинам относят пневмококковую и менингококковую инфекции, развивающиеся по типу молниеносного сепсиса с ДВС крови и кровоизлияниями в надпочечники. Поэтому не позднее, чем за две недели до операции рекомендовано введение пневмококковой, менингококковой и вакцины против Haemophilus influenzae и длительный, не менее 2 лет, профилактический приём бензилпенициллина после спленэктомии. Некоторые авторы предлагают ограничиться введением бициллина-5* (бензатина бензилпенициллин + бензилпенициллин прокаин) ежемесячно в течение 6 мес после операции.
Возможной альтернативой спленэктомии является эндоваскулярная окклюзия селезёнки, проведение которой возможно и у пациентов с глубокой тромбоцитопенией. Для достижения стойкого клинико-гематологического эффекта необходимо поэтапное выключение 90-95% паренхимы органа. Иммунологическая реактивность организма после проведения эндоваскулярной окклюзии селезёнки сохраняется за счёт функционирования 2-5% селезёночной ткани, сохраняющих кровоснабжение за счёт коллатералей, что важно в педиатрической практике. Возможно использование проксимальной эндоваскулярной окклюзии селезёнки за несколько дней до спленэктомии с целью уменьшения риска операции.
Плазмаферез
У пациентов с персистирующей тромбоцитопенией и жизнеугрожающими кровотечениями, несмотря на медикаментозное вмешательство и проведение спленэктомии, возможно использование реинфузии плазмы, пропущенной через колонки с протеином А для быстрого удаления антитромбоцитарных антител. У пациентов с тяжёлой ИТП при этом ускоряется элиминация циркулирующего антитромбоцитарного фактора.
Лечение детей с жизнеугрожающими кровотечениями:
• трансфузии тромбоцитов;
• солюмедрол 500 мг/м2 в сутки внутривенно в 3 введения;
• внутривенно иммуноглобулин 2 г/кг на курс;
• немедленная спленэктомия.
Данные мероприятия могут быть произведены по отдельности или в комбинации в зависимости от тяжести и ответа на лечение.
Прогноз у детей с идиопатической тромбоцитопенической пурпурой
• У 70-80% пациентов ремиссия наступает в течение 6 мес, у 50% — в течение 1 мес от начала заболевания.
• Наступление спонтанной ремиссии после года заболевания нехарактерно, но может быть отмечено даже через несколько лет.
• Прогноз заболевания не зависит от пола, тяжести инициального состояния и обнаружения эозинофилии в костном мозге.
• При выявлении причины ИТП прогноз зависит от её устранения.
• Состояние приблизительно 50-60% пациентов с хронической ИТП стабилизируется без какого-либо лечения и спленэктомии.
Вторичная тромбоцитопеническая пурпура
Как было сказано ранее, тромбоцитопения может быть идиопатической или вторичной в результате ряда известных причин. Вторичная тромбоцитопения, в свою очередь, может быть разделена в зависимости от количества мегакариоцитов.
Дефицит тромболоэтина
Редкой врождённой причиной хронической тромбоцитопении с появлением многочисленных незрелых мегакариоцитов в костном мозге является дефицит тромбопоэтина.
Лечение состоит в трансфузиях плазмы от здоровых доноров или пациентов с ИТП, которое приводит к повышению количества тромбоцитов и появлению признаков созревания мегакариоцитов, либо заместительного применения тромбопоэтина.
Наследственные тромбоцитопении
СЦЕПЛЕННЫЕ С ПОЛОМ ТРОМБОЦИТОПЕНИИ
Тромбоцитопения со сцепленным с полом типом наследования
Тромбоцитопения со сцепленным с полом типом наследования обнаружена исследователями в нескольких семьях. Присутствие нормального количества мегакариоцитов в костном мозге наводит на мысль о том, что тромбоцитопения происходит вследствие сокращения продолжительности жизни тромбоцитов, вызванного их внутренним дефектом. Пациенты плохо отвечают на применение ГК, однако, проведение спленэктомии в некоторых случаях приводит к полной ремиссии.
Синдром Вискотта-Олдрича
Синдром Вискотта-Олдрича проявляется экземой, рецидивирующими инфекциями и тромбоцитопенией. Признаки заболевания появляются в неонатальном периоде или в первые месяцы жизни. Чаще всего дети умирают в раннем возрасте. В неонатальном периоде кровотечения часто представлены меленой, позднее присоединяется пурпура. Тромбоцитопения связана с укорочением жизни тромбоцитов, вызванным их внутренним дефектом, и с нарушением продукции в костном мозге. Проявления заболевания усиливаются при рецидивирующих гнойных инфекциях, включая отит, пневмонию, поражение кожи. У пациентов снижена резистентность и к небактериальным инфекциям, включая вирус простого герпеса и пневмонию, вызванную Pneumocystis carinii.
Гематологические проявления:
• тромбоцитопения (количество тромбоцитов около 30 000/мм3);
• анемия (вызвана потерей крови);
• лейкоцитоз (вызван инфекциями);
• нормальное или повышенное количество мегакариоцитов;
• отсутствие изогемагглютининов, снижение уровня IgM, нормальный или повышенный уровень IgG и IgA;
• в некоторых случаях дефект клеточного иммунитета.
Лечение
• Аллогенная ТКМ, особенно у пациентов с низкой или дискордантной экспрессией белка WASP, по возможности, в первые 5 лет жизни.
• Спленэктомия в тяжёлых случаях у детей старше 5 лет (при невозможности проведения ТКМ) в связи с повышенным риском неконтролируемых инфекций после операции.
• Поддерживающие курсы ВВИГ.
• Трансфузии тромбоцитов при геморрагических проявлениях.
• Назначение ГК-эффекта на тромбоцитопению не оказывают, применяют при экземе.
АУТОСОМНО-НАСЛЕДУЕМЫЕ ТРОМБОЦИТОПЕНИИ
Это гетерогенная группа заболеваний, включающая в себя и доминантные и рецессивные формы тромбоцитопении как моносиндрома, и доминантную аномалию Мея-Хегглина.
В случае тромбоцитопении как моносиндрома описаны семьи как с доминантным, так и с рецессивным типом наследования. Исследование костного мозга обычно позволяет выявить нормальное количество мегакариоцитов при сокращении продолжительности жизни тромбоцитов, связанное с их внутренним дефектом.
Аномалия Мея-Хегглина включает;
• аутосомно-доминантный тип наследования;
• тромбоцитопению различной степени выраженности;
• наличие гигантских тромбоцитов с нормальной функцией и продолжительностью жизни;
• наличие телец Деле в цитоплазме гранулоцитов;
• нормальное количество мегакариоцитов в костном мозге;
• большинство пациентов не имеют симптомов заболевания, у некоторых отмечают геморрагические проявления. В периоде новорождённости кровотечения отсутствуют.