Анемия — состояние гипоксемии, связанное со снижением числа циркулирующих эритроцитов и/или нарушением их способности восполнять потребности тканей в кислороде. Основным маркёром анемии служит концентрация гемоглобина (Hb), осуществляющего доставку кислорода к тканям. Дополнительные маркёры анемии:
• сродство гемоглобина к кислороду;
• внутрисосудистый объём потребления кислорода;
• частота сердечных сокращений;
• удельный объём сердца;
• артериальная оксигенация.
Границей между нормой и патологической анемией принято считать показатель концентрации гемоглобина менее 110 г/л.
Если у пациента есть признаки анемии, то в первую очередь необходимо установить её причину: вызвано ли заболевание патологическими изменениями одной клеточной линии (например, эритроцитов) или нескольких клеточных линий (например, эритроцитов, лейкоцитов и тромбоцитов). Нарушения 2 или 3 клеточных линий обычно указывают на:
• вовлечение в патологический процесс костного мозга (АА, лейкоз);
• заболевания иммунной системы (патологию соединительной ткани, синдром приобретённого иммунодефицита — СПИД);
• деструкцию клеток на периферии (иммунную нейтропению, ИТП);
• иммунную гемолитическую анемию как на самостоятельное заболевание или в сочетании с другой патологией;
• секвестрацию клеток (гиперспленизм).
Ниже приведена этиологическая классификация анемий, в табл. 15.1 представлены их диагностические критерии.
Этиологическая классификация анемий
Нарушения образования эритроцитов:
• дефицит макроэлементов и витаминов:
❖ алиментарный дефицит железа (например, белково-железодефицитная анемия);
❖ повышенная потребность, например, рост (железо), гемолиз (фолиевая кислота);
❖ снижение абсорбции:
- специфическое — недостаточность внутреннего фактора (витамин В12);
- генерализованное — синдром мальабсорбции (например, фолиевая кислота, железо);
❖ повышенная потеря крови:
- острая (железо);
- хроническая — кишечные кровотечения (железо);
• дефицит железа;
• дефицит фолатов;
• дефицит витамина В12;
• дефицит витамина С;
• белковая недостаточность;
• дефицит витамина В6;
• недостаточность тироксина;
• заболевания костного мозга:
❖ патология одной клеточной линии:
- мегакариоцитопения:
- амегакариоцитарная тромбоцитопеническая пурпура;
- дефекты предшественников эритроцитов:
- врождённая эритроцитарная аплазия (АДБ);
- приобретённая эритроцитарная аплазия (транзиторная эритробластопения детей);
- дефекты предшественников лейкоцитов (не связано с анемией):
- врождённая нейтропения;
❖ патология всех клеточных линий (АА, характеризующаяся панцитопенией и гипоклеточным костным мозгом):
- конституциональная:
- АФ;
- семейная анемия без аномалий;
- врождённый дискератоз;
- приобретённая:
- идиопатическая;
- вторичная;
❖ инфильтрация костного мозга:
- de novo (например, лейкоз);
- вторичная (например, нейробластома, лимфома);
• дизэритропоэтическая анемия (подавление эритропоэза, снижение утилизации железа):
❖ инфекция;
❖ патология почек и заболевания печени;
❖ диссеминированные злокачественные новообразования;
❖ заболевания соединительной ткани.
Потеря крови:
• острая постгеморрагическая анемия;
• хроническая постгеморрагическая анемия.
Гемолитическая анемия:
• внутриклеточный гемолиз:
❖ патологические изменения мембран эритроцитов (сфероцитоз, эллиптоцитоз);
❖ патологические изменения активности ферментов эритроцитов (пируваткиназы, Г-6-ФД, глютатионредуктазы);
❖ дефекты гемоглобина:
- гема;
- глобина:
- качественные (например, серповидные клетки);
- количественные (например, талассемия);
• внеклеточный гемолиз:
❖ иммунной природы:
- изоиммунный;
- аутоиммунный:
- идиопатическая анемия;
- вторичная анемия:
иммунологические нарушения (например, СКВ); патологические изменения одной клеточной линии (например, эритроцитов);
патологические изменения нескольких клеточных линий (например, лейкоцитов и тромбоцитов);
❖ неиммунной природы (идиопатическая, вторичная анемия):
- микроангиопатическая анемия;
- токсическая анемия.
Таблица 15-1. Основные критерии диагностики анемий у детей
|
Заболевание |
Диагностические критерии |
|
Дефицит железа |
Гипохромные микроцитарные эритроциты, уровень MCV, МСН и МСНС снижены, высокий уровень ROW, снижение концентрации ферритина в сыворотке крови, высокий уровень FEP, гваяковая проба на скрытую кровь в кале положительна |
|
Дефицит фолатов |
Макроцитарные эритроциты, высокий уровень MCV и RDW, мегалобластный костный мозг, низкое содержание фолатов в сыворотке крови и эритроцитах |
|
Дефицит витамина В|? |
Макроцитарные эритроциты, высокий уровень MCV и RDW, мегалобластный костный мозг, низкое содержание витамина В12 в сыворотке крови, сниженная кислотность желудка; положительный тест Шиллинга |
|
Дефицит витамина С |
Клинические признаки авитаминоза (цинга) |
|
Белковая недостаточность |
Квашиоркор (синдром депигментации и отёка подкожной клетчатки) |
|
Дефицит витамина В6 |
Гипохромные эритроциты, сидеробластный костный мозг, высокое содержание ферритина в сыворотке крови |
|
Недостаточность тироксина |
Кретинизм, низкое содержание Т4, высокое содержание ТТГ |
|
Амегакариоцитарная тромбоцитопеническая пурпура |
Синяки и геморрагии на коже конечностей, отсутствие мегакариоцитов, кровотечения из слизистых оболочек, ранний возраст |
|
Врождённая эритроцитарная аплазия (ДЦБ) |
Отсутствие эритроидных предшественников при исследовании костного мозга |
|
Приобретённая эритроцитарная аплазия (транзи- торная эритробластопения у детей) |
Отсутствие эритроидных предшественников — периодическое |
|
Врождённая нейтропения |
Нейтропения, периодические инфекции |
|
Анемия Фанкони |
Множественные врождённые патологии, ломкость хромосом |
|
Семейная анемия без аномалий |
Наследственность, отсутствие врождённых аномалий |
Окончание табл. 15-1
|
Врождённый дискератоз |
Выявление патологических изменений кожи и слизистой оболочки |
|
Идиопатическая анемия |
Нет установленной причины |
|
Вторичная анемия |
Результат воздействия лекарственных препаратов, радиации, домашних токсинов, инфекций; связана с иммунологическим заболеванием |
|
Инфильтрация костного мозга de novo (например, лейкоз) |
Исследование костного мозга: морфология, цитохимия, иммунологические маркёры, цитогенетика клеток, инфильтрирующих костный мозг |
|
Вторичная инфильтрация костного мозга (например, нейробластома, лимфома) |
Исследование костного мозга и ликвора, визуализация скелета, органов грудной и брюшной полости. Биологические маркёры, иммуноцитология, цитогенетика |
|
Дизэритропоэтическая анемия, вызванная инфекцией |
Случаи системных заболеваний |
|
Дизэритропоэтическая анемия, вызванная патологией почек и заболеваниями печени |
Исследование функций почек и печёночные пробы |
|
Дизэритропоэтическая анемия, вызванная диссеминированными злокачественными новообразованиями |
Клинические проявления |
|
Дизэритропоэтическая анемия, вызванная заболеваниями соединительной ткани |
Ревматоидный артрит/СКВ |
|
Острая постгеморрагическая анемия |
Оценка кровопотери, ОЦК, определение концентрации Нb после восстановления ОЦК |
|
Хроническая постгеморрагическая анемия |
Проба Оверта или тест на скрытую кровь |
|
Внутриклеточный гемолиз вследствие патологических изменений мембран эритроцитов (сфероцитоз, эллиптоцитоз) |
Морфологическое исследование крови, кривая Прайс- Джонса, осмотическая резистентность |
|
Внутриклеточный гемолиз вследствие патологических изменений ферментов эритроцитов (пируваткиназы, Г-6-ФД, глутатионредуктазы) |
Аутогемолиз, ферментативный ответ |
|
Качественная анемия, вызванная дефектами гемоглобина (например, серповидные клетки) |
Электрофорез НЬ |
|
Количественная анемия, вызванная дефектами гемоглобина (например, талассемия) |
Содержание Hb F, Нb A2 |
|
Иммунная анемия |
Проба Кумбса |
|
Идиопатическая или вторичная анемия с внеклеточным гемолизом |
Проба Кумбса, выявление антител |
|
Иммунологические нарушения (например, СКВ) |
Снижение С3, С4, СН50, ANA положительный |
|
Иммунологические нарушения одной клеточной линии (например, эритроциты) |
Анемия — проба Кумбса положительная |
|
Иммунологические нарушения нескольких клеточных линий (например, лейкоциты, тромбоциты) |
Нейтропения — иммунная ИТП, тромбоцитопения |
|
Микроангиопатическая анемия |
Внутрисосудистый гемолиз как результат тромбогеморрагического синдрома |
|
Токсическая анемия |
Отравление грибами, укусы ядовитых змей и т.д. |
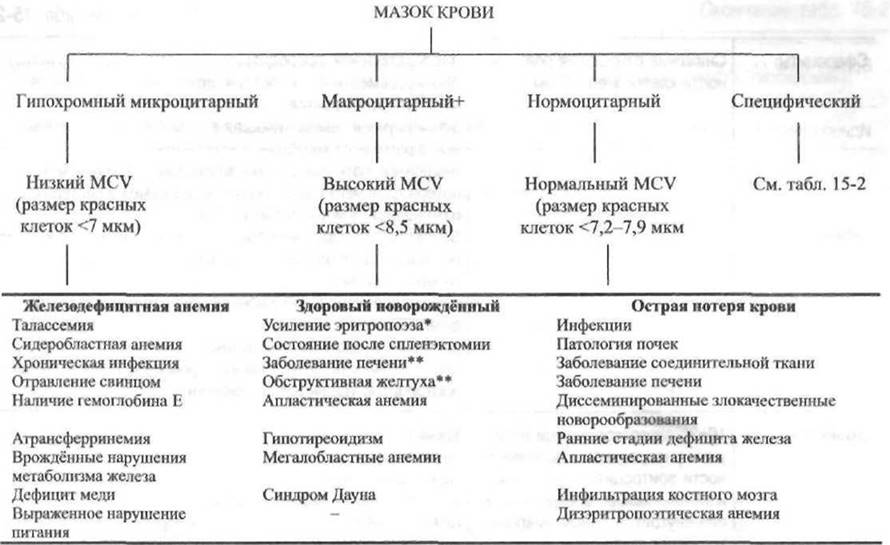
Рис. 15-1. Алгоритм диагностики анемий на основе исследования мазка крови.
+ Ложный макроцитоз (высокий показатель MCV) может быть результатом макроагглютинации красных клеток (например, Mycoplasma pneumoniae и аутоиммунная гемолитическая анемия);
* повышенный уровень ретикулоцитов;
** на основе увеличения площади клеточных мембран, что, в свою очередь, ведёт к увеличению отношения поверхности клетки к её объёму. Площадь мембран увеличивается вследствие изменения липидного обмена красных клеток и липидного баланса при данных патологиях.
ДИАГНОСТИКА
При диагностике анемий очень информативен анализ мазка крови. Результаты анализа указывают на характер анемии (гипохромный, микроцитарный, нормо- или макроцитарный), а также специфические морфологические изменения (например, сфероциты, серповидные клетки, мишеневидные клетки). Средний объём эритроцита (MCV) свидетельствует о размерах красных клеток, определяя, например, микроцитоз (<7 мкм), макроцитоз (>8,5 мкм) или нормоцитоз (7,2-7,9 мкм). На рис. 15-1 представлен алгоритм диагностики анемий при исследовании препаратов крови. В табл. 15-2 приведён метод дифференциальной диагностики анемий, основанный на специфических морфологических изменениях красных клеток.
Таблица 15-2. Специфические нарушения морфологии эритроцитов
|
Тип нарушения |
Определение |
При каких заболеваниях или состояниях встречается |
|
Мишеневидные клетки |
Увеличение показателя отношения поверхности клетки к её объёму |
Талассемия; |
|
гемоглобинопатии: |
||
|
- Нb АС или СС; - Hb SS, SC, S-Thal; - заболевания печени; - состояние после сплензктомии или гипоспления; - выраженный дефицит железа; - Нb Е (гетерозиготный или гомозиготный); - LCAT-недостаточность: - врождённая лецитин-холестерол-ацилтрансферазная недостаточность (помутнение роговицы, протеинурия, мишеневидные клетки, выраженная анемия); - абеталипопротеинемия |
Продолжение табл. 15-2
|
Сфероциты |
Снижение отношения поверхности клетки к её объёму |
Наследственный сфероцитоз; АВО-несовместимость: потеря антительных фрагментов мембран эритроцитов; аутоиммунная гемолитическая анемия: потеря антительных фрагментов мембран эритроцитов; микроангиопатическая гемолитическая анемия (МАНА): потеря фрагмента эритроцита после взаимодействия с изменённой поверхностью клетки; SS-патология: фрагмент эритроцита перемещается в ретикулоэндотелиальную систему; гиперспленизм; ожоги: фрагмент повреждённых эритроцитов удаляется селезёнкой; посттрансфузионные состояния; недостаточность пируваткиназы: фрагмент повреждённых эритроцитов удаляется селезёнкой |
|
Эхиноциты |
10-30 отростков равной величины распределены по поверхности эритроцитов. Вызваны изменениями во внеклеточном или внутриклеточном микроокружении |
Уремия; дегидратация; заболевания печени; недостаточность пируваткиназы; пептическая язва или карцинома желудка; состояние после трансфузии эритроцитов; редкие врождённые анемии вследствие уменьшения содержания внутриклеточного калия |
|
Акантоциты (шпоровидные клетки) |
Клетки с 5-10 отростками различной длины, отростки хаотично расположены, различной толщины с широким основанием; изменённые клетки меньше нормальных, т.к. последние имеют сферическую форму |
Заболевания печени; Диссеминированное внутрисосудистое свёртывание (ДВС) (и другие МАНА); состояние после спленэктомии или гипоспления; дефицит витамина Е; гипотироидизм; абеталипопротеинемия: редкая врождённая патология; 50-100% клеток — акантоциты; сопутствующие нарушения (мальабсорбция жиров, пигментация сетчатки, неврологические нарушения); состояния мальабсорбции |
|
Пикноциты |
Искривлённые, гиперхромные, сморщенные эритроциты; могут быть похожи на зхиноциты и акантоциты |
|
|
Шизоциты |
Клетки шлемовидной, треугольной формы или небольшие фрагменты. Образованы вследствие фрагментации при взаимодействии с патологической поверхностью сосудов (например, фибриновые тяжи, васкулит, искусственные сосуды, участвующие в циркуляции) |
ДВС-синдром; тяжёлая гемолитическая анемия (например, недостаточность Г-6-ФД); микроангиопатическая гемолитическая анемия; гемолитический уремический синдром; протезирование сердечного клапана, патология сердечного клапана, сердечные бляшки, коарктация аорты; заболевания соединительной ткани (например, СКВ); синдром Казабаха-Меррита: молниеносная пурпура; тромбоз почечных вен; ожоги (сферошизоцитоз как результат воздействия высоких температур); тромбоцитарная тромбоцитопеническая пурпура; отторжение пересаженных тканей; уремия, острый тубулярный некроз, гломерулонефрит; злокачественная гипертензия; системный амипокдоз; цирроз печени; диссеминированный канцероматоз; хроническая рецидивирующая шистоцитарная гемолитическая анемия |
Окончание табл. 15-2
|
Эллиптоциты |
Эллиптоидные клетки, нормох- ромные; в норме составляют менее 1 % общего содержания эритроцитов; большие количества редко бывают у здоровых пациентов |
Наследственный эллиптоцитоз; дефицит железа (с нарастанием тяжести, гипохромия); SS-патология; большая талассемия; тяжёлая бактериальная инфекция; малая SA; лейкоэритробластная реакция; мегалобластная анемия; при любой анемии могут присутствовать до 10% эллиптоцитов; малярия |
|
Каплевидные клетки |
Клетки в форме капли, обычно микроцитарные, часто ещё и гипохромные |
Новорождённый; большая талассемия; лейкоэритробластные реакции; миелопролиферативный синдром |
|
Стоматоциты |
Выраженное щелевидное просветление в центре клетки |
Норма (в небольших количествах); наследственный стоматоцитоз; талассемия; острое алкогольное отравление; RhO-патология (отсутствие Rh-комплекса); заболевания печени; злокачественные новообразования |
|
Ядерные эритроциты |
Обнаружение ядерных клеток в периферической крови после первой недели жизни считают патологией |
Новорождённый (первые 3-4 дня жизни); выраженная стимуляция костного мозга; гипоксия (особенно нарушения сердечной деятельности); острое кровотечение; тяжёлая гемолитическая анемия (например, талассемия, SS-гемоглобинопатия); врождённые инфекции (сепсис, врождённый сифилис, цитомегаловирус, краснуха); состояние после спленэктомии или гипоспления: в норме селезёнка удаляет; ядерные эритроциты; лейкоэритробластные реакции: наблюдаются в сочетании с экстрамедуллярным гемопоэзом и замещением костного мозга, чаще при лейкозах и солидных опухолях; может быть при грибковых и микобактериальных инфекциях; каплевидные эритроциты, 10 000-20 000 лейкоцитов (WBCs) с небольшим или значительным числом метамиелоцитов, миелоцитов и промиелоцитов; тромбоцитоз с большими тромбоцитами, содержащими ядра; мегалобластная анемия; дизэритропоэтические анемии |
|
Пузырчатые клетки |
Под мембраной эритроцита присутствует свободный от гемоглобина участок, похожий на пузырь |
Недостаточность Г-6-ФД (во время эпизодов гемолиза); SS-патология; эмболия лёгкого |
|
Базофильные включения |
Грубые или тонкие пятнистые базофильные включения, представляющие агрегаты рибосо- мальной РНК |
Гемолитическая анемия (например, малая талассемия); ЖДА; отравление свинцом |
|
Тельца Говелла-Жолли |
Небольшого размера, чётко распознаваемые, округлые, хорошо окрашенные включения; 1 мкм в диаметре; расположены по периферии клетки |
Постспленэктомия или гипоспления; новорождённый; мегапобластные анемии; дизэритропоэтические анемии; различные виды анемий (редко ЖДА, наследственный сфероцитоз) |
Для дифференциальной диагностики анемий достаточно информативны показатели MCV и уровня ретикулоцитов (рис. 15-2). Повышение числа ретикулоцитов свидетельствует о хронической кровопотере или гемолизе, нормальный или сниженный уровень — о нарушении образования красных клеток крови.
Уровень ретикулоцитов помогает определить степень анемии; ретикулоцитар- ный индекс (см. ниже) — наиболее точный показатель эритропоэза. У пациентов с кровотечением или гемолизом ретикулоцитарный индекс составляет около 3%,
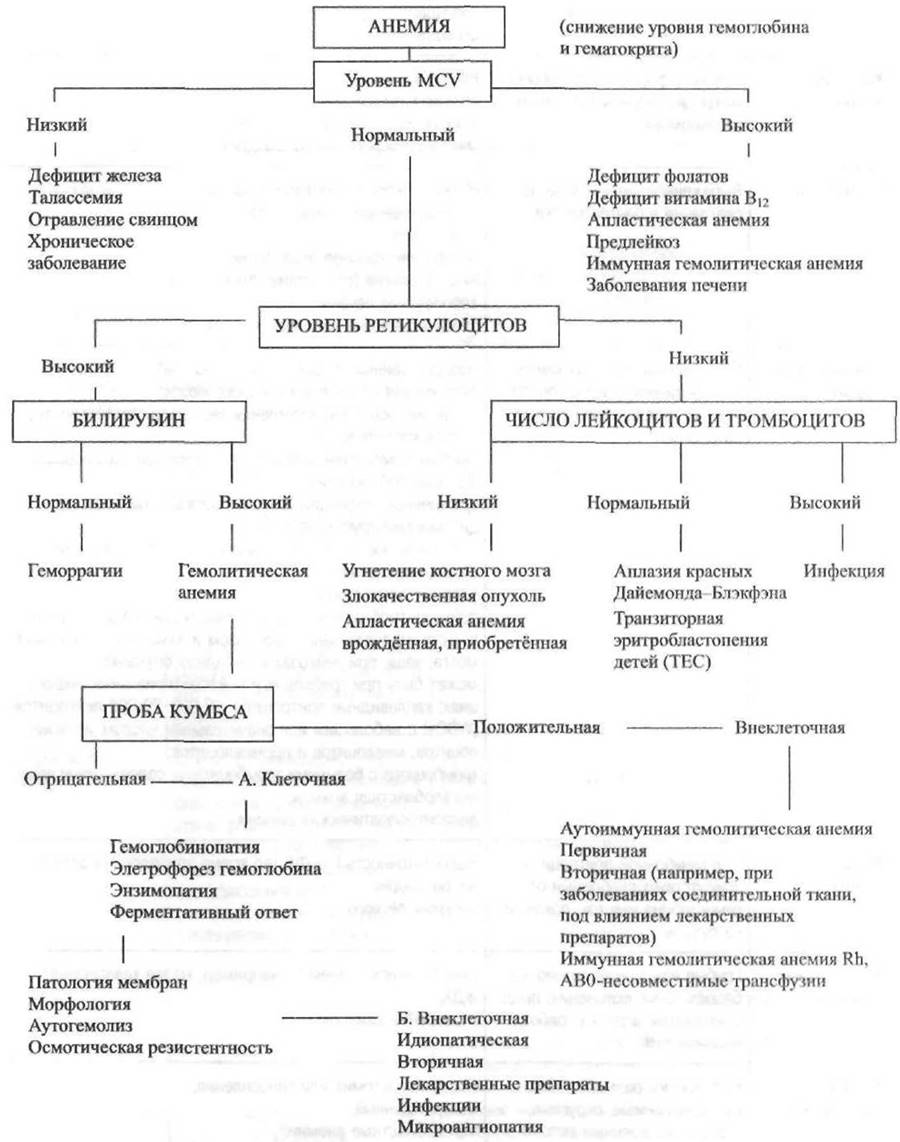
Рис. 15-2. Алгоритм диагностики анемий на основе данных показателей среднего объёма эритроцитов и уровня ретикулоцитов.
тогда как у больных с анемией вследствие снижения продукции эритроцитов этот индекс менее 3%, а чаще — менее 1,5%.
Показатели MCV и RDW, определяемые при исследовании крови на автоматическом анализаторе, помогают установить морфологию и природу анемий, что, в свою очередь, привело к разработке классификации, основанной на данных этих индексов (табл. 15-3).
Таблица 15-3. Классификация анемий на основании показателей среднего объёма эритроцита и ширины распределения эритроцитов по объёму
|
RDW |
Уровень MCV |
||
|
низкий |
нормальный |
высокий |
|
|
Нормальная |
Микроцитарная гомогенная |
Нормоцитарная гомогенная |
Макроцитарная гомогенная |
|
Нормальная |
Гетерозиготная талассемия Хронические заболевания |
Норма Хроническое заболевание Хроническое заболевание печени Неанемическая гемоглобинопатия (например, AS, АС) Химиотерапия Хронический миелоцитарный лейкоз Геморрагия Наследственный сфероцитоз |
АА Предлейкоз |
|
Высокая |
Микроцитарная гетерогенная |
Нормоцитарная гетерогенная |
Макроцитарная гетерогенная |
|
Высокая |
Дефицит железа S β-талассемия Нb Н Фрагментация эритроцитов |
Ранние стадии недостаточности железа или фолатов Смешанные недостаточности Гемоглобинопатии (например, SS, SC) Миелофиброз Сидеробластная анемия |
Дефицит фолатов Дефицит витамина В12 Иммунная гемолитическая анемия Холодовые агглютинины |
В более сложных случаях проводят пункцию костного мозга с окрашиванием препарата для определения запасов железа и выявления признаков сидеробластной анемии. Исследования костного мозга позволяют определить нормобластную, межобластную или сидеробластную морфологию. На рис. 15.3 представлены возможные причины каждой из них.
Ретикулоцитарный индекс = Число ретикулоцитов х (Гематокрит больного/Гематокрит нормальный).
Например, количество ретикулоцитов соответствует 6%, гематокрит — 15%:
Ретикулоцитарный индекс = 6 х (15/45) = 2%.
Диагностическое обследование включает следующие этапы:
• детальное изучение истории болезни и физикальное обследование больного (см. табл. 15-1);
• полный клинический анализ крови для установления причины анемии: вызвана ли она патологическими изменениями только одной клеточной линии (эритроциты) или 3 клеточных линий (эритроциты, лейкоциты, тромбоциты);
• определение морфологических характеристик анемий на основании исследования мазка крови (см. табл. 15-2), показателей MCV (см. рис. 15-1, 15-2) и RDW (см. табл. 15-3), а также морфологии лейкоцитов и тромбоцитов;

Рис. 15-3. Причины нормо-, мегало- и сидеробластной морфологии костного мозга.
• исследование аспирата костного мозга (проводят по мере необходимости) для определения эритроидной, миелоидной или мегакариоцитарной морфологии, определяющей тип эритропоэза (нормо-, мегало- или сидеробластный), а также для исключения патологии со стороны костного мозга (например, АА, лейкоза, доброкачественной или злокачественной инфильтрации костного мозга, рис. 15-3);
• установление более глубоких причин анемий с помощью дополнительных тестов. Лабораторные методы исследования, используемые при диагностике анемий:
• первичные методы исследования:
❖ определение уровня НЬ и гематокрита;
❖ определение количества эритроцитов и их индексов, включая MCV и RDW;
❖ определение количества ретикулоцитов; о изучение окрашенного мазка крови;
❖ определение количества лейкоцитов и их предшественников;
❖ определение количества тромбоцитов;
• предполагаемый дефицит железа:
• свободные протопорфирины эритроцитов;
❖ уровень ферритина сыворотки крови;
❖ исследование кала на скрытую кровь;
• сканирование путём внутривенного введения 99mТс-меченых эритроцитов для обнаружения меккелева дивертикула;
❖ эндоскопия (восходящей и нисходящей кишки);
• предполагаемый дефицит витамина В12 или фолиевой кислоты:
❖ исследование костного мозга;
❖ уровень витамина В12 в сыворотке крови; о уровень фолатов сыворотки крови;
❖ исследование желудка после введения гистамина;
❖ тест на абсорбцию витамина В12 (радиоактивный кобальт, тест Шиллинга);
• предполагаемая гемолитическая анемия:
❖ признаки поломок эритроцитов:
- анализ мазка крови;
- уровень билирубина сыворотки крови;
- экскреция уробилиногена мочи;
- гаптоглобин сыворотки крови;
❖ признаки регенерации эритроцитов:
- анализ мазка крови;
- уровень ретикулоцитов;
- рентген скелета;
❖ признаки гемолитической анемии:
- анализ мазка крови;
- тест на осмотическую резистентность;
- тест на аутогемолиз;
- исследование НЬ:
- серповидный тест;
- электрофорез НЬ;
- определение НЬ F — мазок по Клейхауеру-Бетке;
- тест на нагревание;
- исследование ферментов:
- исследование телец Гейнца;
- ферментативный анализ;
❖ признаки внеклеточной иммунной гемолитической анемии:
- антиглобулиновый тест;
- тест на лизис кислот сыворотки крови;
- сахарозный гемолитический тест;
- антитела Доната-Ландштейнера;
- антиядерные антитела (ANA);
• предполагаемая АА или лейкоз:
❖ аспирация и биопсия костного мозга — цитохимия, иммунологические маркёры;
❖ хромосомный анализ;
❖ рентген скелета;
• другие тесты, обычно применяемые при диагностике первичной патологии:
❖ серология вирусов, например, вирус иммунодефицита человека (ВИЧ);
❖ определение ANA, комплемента, СН50;
❖ тест на содержание крови в моче;
❖ определение концентрации креатинина;
❖ биопсия тканей (кожи, лимфатических узлов, печени).
Железодефицитная анемия
Описано 3 железодефицитных состояния (ЖДС):
• прелатентный дефицит железа;
• латентный дефицит железа;
• ЖДА.
При прелатентном дефиците железа содержание железа снижено только в депо при сохранении транспортного и гемоглобинового фондов. Отсутствие клинических проявлений и чётких критериев диагностики позволяют не придавать этому состоянию практического значения. Латентный дефицит железа, составляющий 70% всех ЖДС, считают не болезнью, а функциональным расстройством с отрицательным балансом железа, оно не имеет самостоятельного кода по МКБ-10. При латентном дефиците железа наблюдается характерная клиническая картина: сидеропенический синдром, но содержание Нb остаётся в пределах нормальных значений, что не позволяет выявить лиц с этим состоянием из общей популяции по этому лабораторному параметру. ЖДА (код по МКБ-10 — D50) — болезнь, самостоятельная нозологическая форма, составляющая 30% всех ЖДС. При этом заболевании выявляют:
• анемический и сидеропенический синдромы;
• снижение концентрации Нb и сывороточного железа (СЖ);
• повышение общей железосвязывающей способности сыворотки (ОЖСС);
• снижение концентрации сывороточного ферритина (СФ).
ЭПИДЕМИОЛОГИЯ
Материалы, опубликованные ВОЗ (1998), позволяют осознать важность проблемы дефицита железа (ДЖ): в мире 1,8 млрд человек страдают ЖДА.
ЖДА может быть названа социально значимым заболеванием. Распространённость ЖДА у детей в возрасте 2,5 лет в Нигерии составляет 56%, в России — 24,7%, в Швеции — 7%. По мнению экспертов ВОЗ, если распространённость ЖДА превышает 30%, эта проблема выходит за рамки медицинской и требует принятия решений на государственном уровне.
По данным официальной статистики М3 РФ, в России наблюдается значительный рост заболеваемости анемией детей и подростков.
В детском возрасте 90% всех анемий составляют ЖДА. Таким образом, назначая приём препаратов железа при всех анемиях, врач «угадает» в 9 случаях из 10. Остальные 10% анемий включают врождённые и приобретённые гемолитические и АА, а также анемии при хронических болезнях.
В норме организм взрослого здорового человека содержит около 3-5 г железа, таким образом, железо может быть отнесено к микроэлементам. Железо распределено в организме неравномерно. Приблизительно 2/3 железа содержится в НЬ эритроцитов — это циркулирующий фонд (или пул) железа. У взрослых этот пул составляет 2-2,5 г, у доношенных новорождённых — 0,3-0,4 г, и у недоношенных новорождённых — 0,1-0,2 г. Относительно много железа содержится в миоглобине: 0,1 г — у мужчин и 0,05-0,07 г — у женщин. В организме человека содержится более 70 белков и ферментов, в состав которых входит железо (например, трансферрин, лактоферрин), суммарное количество железа в них составляет 0,05-0,07 г. Железо, переносимое транспортным белком трансферрином, составляет около 1% (транспортный фонд железа). Для медицинской практики чрезвычайно важны запасы железа (депо, запасной фонд), составляющие около 1/3 всего железа в организме человека. Функцию депо выполняют следующие органы:
• печень;
• селезёнка;
• костный мозг;
• головной мозг.
Железо содержится в депо в виде ферритина. Количество железа в депо может быть охарактеризовано с помощью определения концентрации СФ. На сегодняшний день СФ — единственный международно-признанный маркёр запасов железа. Конечный продукт обмена железа — гемосидерин, откладывающийся в тканях.
Железо — важнейший кофактор ферментов митохондриальной дыхательной цепи, цитратного цикла, синтеза ДНК, оно играет важную роль в связывании и транспорте кислорода Нb и миоглобином; белки, содержащие железо, необходимы для метаболизма коллагена, катехоламинов, тирозина. Вследствие каталитического действия железа в реакции Fe2+↔Fe3+свободное нехелатированное железо образует гидроксильные радикалы, способные вызвать повреждение клеточных мембран и гибель клеток. В процессе эволюции защита от повреждающего действия свободного железа была решена с помощью образования специализированных молекул для абсорбции железа из пищи, его всасывания, транспорта и депонирования в нетоксичной растворимой форме. Транспорт и депонирование железа осуществляются специальными белками: трансферрином, трансферриновым рецептором, ферритином. Синтез этих белков регулируется с помощью специального механизма и зависит от потребностей организма.
Метаболизм железа у здорового человека замкнут в цикл. Ежедневно человек теряет около 1 мг железа с биологическими жидкостями и слущенным эпителием ЖКТ. Ровно столько же способно всасываться в ЖКТ из продуктов питания. Следует чётко представлять, что железо поступает в организм только с продуктами питания. Таким образом, ежедневно 1 мг железа теряется и 1 мг всасывается. В процессе разрушения старых эритроцитов освобождается железо, которое утилизируется макрофагами и вновь используется при построении гема. В организме существует специальный механизм всасывания железа, однако выводится оно пассивно, то есть физиологического механизма выведения железа не существует. Следовательно, если всасывание железа из пищи не удовлетворяет потребностям организма, ДЖ возникает независимо от причины.
ЭТИОЛОГИЯ
Известно более 10 видов нарушений обмена железа, приводящих к развитию ЖДС. Наибольшее значение имеют:
• ДЖ в пище, имеющий значение в развитии ЖДС как у детей от самого раннего до подросткового возраста, так и у взрослых и людей пожилого возраста;
• нарушение всасывания железа в двенадцатиперстной кишке и верхних отделах тонкой кишки в результате воспаления, аллергического отёка слизистой оболочки, лямблиоза, инфицированности Helicobacterjejuni, при кровотечениях;
• нарушение перехода Fe3+→Fe2+вследствие дефицита андрогенов, аскорбиновой кислоты, атрофического гастрита, приводящих к недостаточному образованию гастроферрина.
ДИАГНОСТИКА
В соответствии с рекомендациями ВОЗ, стандартизованы следующие критерии диагностики ЖДА:
• снижение уровня СЖ менее 12 мкмоль/л;
• повышение ОЖСС более 69 мкмоль/л;
• насыщение трансферрина железом менее 17%;
• содержание Нb ниже 110 г/л в возрасте до 6 лет и ниже 120 г/л — в возрасте старше 6 лет.
Таким образом, ВОЗ рекомендует достаточно точные критерии для диагностики ЖДА, однако методы диагностики требуют забора крови из вены и проведения достаточно дорогих биохимических исследований, что не всегда возможно в российских лечебных учреждениях. Имеются попытки минимизации критериев диагностики ЖДА.
Федеральная служба по заболеваемости, эпидемиологии и образованию (United States Federal Government Centers for Disease Control — CDC) со штаб-квартирой в Атланте (Джорджия, США) рекомендует использовать для диагностики ЖДА 2 доступных критерия: снижение концентрации Нb и гематокрита (Ht) при отсутствии у больного других заболеваний. Устанавливают предположительный диагноз ЖДА и назначают лечение препаратами железа на 4 нед из расчёта 3 мг элементарного железа на 1 кг массы тела больного в сутки. Достоинство данных рекомендаций - регистрация ответа на терапию препаратами железа по строго фиксированным критериям. К концу 4-й нед лечения концентрация Нb должна повыситься на 10 г/л по отношению к исходной, a Ht — на 3%. Такой ответ подтверждает диагноз «ЖДА», и лечение продолжают в течение нескольких месяцев. Если ответ не получен, рекомендуют остановить лечение препаратами железа и пересмотреть данный случай с точки зрения диагностики процесса. Перегрузка железом организма за 4 нед при приёме препаратов железа внутрь маловероятна.
Лабораторную диагностику ЖДА осуществляют с помощью:
• общего анализа крови, выполненного «ручным» методом;
• анализа крови, выполненного на автоматическом анализаторе крови;
• биохимических исследований.
При диагностике любой анемии обязательно выполнение общего анализа крови с определением количества ретикулоцитов. Врач ориентируется на гипохромный и микроцитарный характер анемии. В общем анализе крови, выполненном «ручным» методом, выявляют:
• снижение концентрации Нb (<110 г/л);
• нормальное или сниженное (<3,8х1012/л) количество эритроцитов;
• снижение цветового показателя (<0,76);
• нормальное (реже слегка повышенное) содержание ретикулоцитов (0,2-1,2%);
• увеличение скорости оседания эритроцитов (СОЭ) (>12-16 мм/ч);
• анизоцитоз (характерны микроциты) и пойкилоцитоз эритроцитов.
Ошибка определения параметров может достигать 5% и более. Стоимость одного общего анализа крови составляет около 5 долларов США.
Точным и удобным методом диагностики и дифференциальной диагностики служит метод определения эритроцитарных показателей на автоматических анализаторах крови. Исследование проводят как в венозной, так и в капиллярной крови. Ошибка в определении параметров значительно ниже, чем при «ручном» методе, и составляет менее 1%. При развитии ДЖ раньше всего повышается показатель выраженности анизоцитоза эритроцитов — RDW (норма <14,5%). С помощью определения MCV регистрируют микроцитоз (норма — 80-94 фл). Кроме того, снижается среднее содержание Нb в эритроците — МСН (норма — 27-31 пг) и средняя концентрация Нb в эритроците — МСНС (норма — 32-36 г/л). Стоимость одного анализа, выполненного на автоматическом гематологическом анализаторе, составляет около 3 долларов США.
Биохимические показатели, подтверждающие ДЖ в организме, информативны, однако требуют забора крови из вены и достаточно дороги (стоимость однократного определения СЖ, ОЖСС, СФ составляет более 33 долларов США). Наиболее важным критерием ДЖ считают снижение концентрации СФ (<30 нг/мл). Однако ферритин — белок острой фазы воспаления, его концентрация на фоне воспаления или беременности может быть повышена и «замаскирует» имеющийся ДЖ. Необходимо иметь в виду, что показатель СЖ нестабилен, так как содержание железа в организме подвержено колебаниям, имеющим суточный ритм, и зависит от диеты. Насыщение трансферрина железом — расчётный коэффициент, определяемый по формуле:
(СЖ/ОЖСС) х 100%.
Трансферрин не может быть насыщен железом более чем на 50%, что обусловлено его биохимической структурой, чаще всего насыщение составляет от 30 до 40%. При падении насыщения трансферрина железом ниже 16% эффективный эритропоэз невозможен.
Основные причины развития ЖДА у детей и подростков:
• алиментарный ДЖ вследствие несбалансированного питания;
• ДЖ при рождении;
• повышенные потребности организма в железе вследствие бурного роста ребёнка;
• потери железа, превышающие физиологические.
И.Я. Конь (2001) приводит 3 основных алиментарно-зависимых фактора в развитии ДЖ у детей:
• сниженное поступление железа с пищей;
• сниженное всасывание;
• увеличенные потери.
Рассматривают следующие причины сниженного поступления железа с пищей:
• отсутствие грудного вскармливания;
• использование в питании детей раннего возраста частично адаптированных и неадаптированных молочных смесей, не обогащённых железом каш;
• позднее введение прикорма;
• сниженное потребление витамина С и др.
К понижению всасывания железа приводит использование в питании большого количества растительных волокон, избыток белка, кальция, полифенолов. Увеличенные потери железа возможны при раннем введении в питание ребёнка цельного молока и кефира, что приводит к появлению диапедезных кровотечений из желудка и тонкой кишки и потерям Нb путём экскреции с калом.
Для профилактики ДЖ по-прежнему важна работа по повышению распространённости грудного вскармливания. В грудном молоке содержится железо с самой высокой биологической доступностью — 50%, что не имеет аналогов.
В рационе человека выделяют гемовые и негемовые продукты питания; негемовые продукты питания преобладают (90%), гемовые составляют около 10%. Степень усвоения железа из этих видов продуктов питания также различна. Усвоение железа из риса, кукурузы, сои, бобов, фасоли, шпината, муки составляет 1-7% его содержания в продукте. Усвоение железа из мясных продуктов составляет от 18-20 до 30%.
Многолетнее питание продуктами растительного происхождения — поставщиками трудноусвояемого негемового железа — и отказ от мясных продуктов, богатых легкоусвояемым гемовым железом, может приводить к ЖДА. Это подтверждается осмотром вегетарианцев. «Цивилизованные» вегетарианцы западных стран обязательно применяют поливитамины, микроэлементы, в том числе и препараты железа на фоне растительной диеты, что и позволяет им иметь нормальный уровень НЬ.
Анемия беременных обычно вызвана 2 причинами: отрицательным балансом железа в организме и его недостаточным поступлением. ДЖ в организме беременной опасен многочисленными рисками для неё самой и для плода, в частности:
• плацентарной недостаточностью;
• внутриутробной гибелью плода;
• выкидышами;
• преждевременными родами;
• низкой массой тела ребёнка при рождении;
• преэклампсией;
• пиелонефритом;
• послеродовыми инфекциями;
• кровоточивостью.
Потребности беременной в железе настолько возрастают, что не могут быть покрыты с помощью обычной диеты, даже при всасывании железа, возросшем в несколько раз. Общие затраты железа беременной складываются из:
• дополнительных эритроцитов матери — 450 мг;
• тканей плода, плаценты и пуповины — 360 мг;
• потери крови в процессе родов — 200-250 мг;
• ежесуточной потери через ЖКТ и с потом — 1 мг;
• потери с молоком при кормлении грудью — 1 мг.
Суммарные потери железа составляют более 1000 мг.
Критериями анемии у беременных считают снижение концентрации НЬ менее ПО г/л в I и III триместрах беременности и менее 105 г/л — во II триместре.
Как известно, концентрация НЬ у 30% женщин после родов ниже 100 г/л, а у 10% женщин — ниже 80 г/л, что соответствует анемии средней степени тяжести, требующей лечения и усугубляющейся в связи с периодом лактации. Причины развития послеродовой анемии у женщин:
• истощение запасов железа в депо в процессе беременности;
• потеря крови во время родов.
Потеря крови при физиологически протекающих родах составляет 400-500 мл (200-250 мг железа), а при многоплодной беременности или кесаревом сечении повышается до 900 мл (450 мг железа). Традиционные методы лечения послеродовой анемии:
• переливание эритроцитарной массы в тяжёлых случаях, требующих неотложного лечения;
• применение препаратов железа для приёма внутрь в случаях нетяжёлой анемии.
Применение внутривенных препаратов железа при лечении послеродовой анемии оказалось эффективным и быстрым методом лечения. Это чрезвычайно важно в связи с тем, что женщин рано выписывают из родильного дома и им предстоит период лактации, требующий дополнительно не менее 1 мг железа в сутки. Как показали результаты исследований, применение препарата венофер* [железа (III) гидроксид сахарозный комплекс; 3 внутривенных введения по 200 мг в течение недели] приводит к революционному результату: в группе из 30 женщин отмечено повышение средней концентрации Нb с 70,7 до 109,3 г/л. Таким образом, продемонстрирован переход тяжёлой анемии в лёгкую в рекордно короткие сроки. Подобное лечение служит альтернативой гемотрансфузиям. В нашей стране такие же результаты получены группой сотрудников под руководством профессора Р.И. Шалиной (2003).
Хроническую постгеморрагическую анемию, связанную с длительной потерей небольшого объёма крови, также относят к ЖДА и лечат по принципам лечения ЖДА. При лечении хронической постгеморрагической анемии прежде всего необходимо обнаружить источник кровопотери и устранить его. Для пациентов мужского пола более характерны потери из ЖКТ, вызванные:
• язвенными кровотечениями;
• полипами толстой кишки;
• неспецифическим язвенным колитом;
• ангиоматозом кишечника;
• наличием меккелева дивертикула;
• опухолями желудка и кишечника (у взрослых);
• кровотечениями из геморроидальных образований (у взрослых).
У пациентов женского пола на первом месте стоят кровотечения, связанные с ювенильными маточными кровотечениями у девушек пубертатного возраста и длительными и обильными менструациями, отмечаемыми у 12-15% женщин репродуктивного возраста. Потери Нb из ЖКТ занимают у женщин второе место.
Доноры, часто сдающие кровь (регулярные доноры), находятся в группе риска развития ЖДС или уже имеют ЖДА. Преодоление ДЖ у доноров возможно с помощью:
• перерывов в сдаче крови (не менее 3 мес);
• полноценного питания;
• назначения препаратов железа для приёма внутрь.
Единственный недостаток этих рекомендаций — необходимость их длительного выполнения. Быстрое преодоление ДЖ у регулярных доноров принципиально возможно с помощью введения внутривенных препаратов железа, например, с помощью зарегистрированного в нашей стране препарата венофер*. Для этого имеются следующие обоснования:
• венозный доступ при заборе крови обеспечен;
• объём кровопотери известен;
• величину потери железа из организма рассчитывают исходя из объёма сданной крови (одномоментная эксфузия 500 мл цельной крови приводит к потере 250 мг железа).
При этом увеличивается себестоимость цельной крови и её компонентов, однако необходимо, прежде всего, учитывать самочувствие донора, снижение качества его жизни в период преодоления ЖДА. Вполне возможно, что использование внутривенных препаратов железа позволит донорам чаще сдавать кровь, что важно при существующем дефиците доноров.
Дифференциальная диагностика
Дифференциальную диагностику ЖДА следует проводить с анемией при хронических болезнях и анемиями, вызванными дефицитом фолиевой кислоты или витамина В12, то есть внутри группы «дефицитных» анемий. Анемия при хронических болезнях — самостоятельная нозологическая форма, имеющая код по МКБ-10 — D63.8. Основные причины развития анемии при хронических болезнях:
• наличие основного хронического заболевания (как правило, известного врачам!);
• инфекции, протекающие хронически (туберкулёз, сепсис, остеомиелит);
• системные заболевания соединительной ткани (ревматоидный артрит, СКВ);
• хронические заболевания печени (гепатиты, цирроз);
• злокачественные новообразования.
Патогенез развития анемии при хронических болезнях окончательно неясен, однако известны следующие механизмы:
• нарушение метаболизма железа при его достаточном количестве в организме, при этом затруднено использование железа и реутилизация его из макрофагов;
• гемолиз эритроцитов;
• супрессия эритропоэза ингибиторами (средние молекулы, продукты пере- кисного окисления липидов, цитокины, ФИО, ИЛ-1, замещение опухолевыми клетками;
• неадекватная продукция эритропоэтина: повышение его выработки в ответ на анемию, однако его количество недостаточно для компенсации анемии.
Лабораторные критерии диагностики анемии при хронических болезнях:
• снижение концентрации НЬ (нерезкое);
• снижение количества эритроцитов (нерезкое);
• микроцитарный характер анемии;
• норморегенераторный характер анемии;
• снижение СЖ;
• снижение ОЖСС (!);
• нормальное или повышенное (!) содержание СФ;
• увеличение СОЭ.
ЛЕЧЕНИЕ
Принципы лечения
Препараты железа — основные лекарства для лечения ЖДА, они представлены многочисленными формами препаратов железа для приёма внутрь (капли, сироп, таблетки).
В табл. 15-4 представлены современные препараты железа для приёма внутрь, зарегистрированные и доступные в России. Для расчёта необходимого количества препарата необходимо знать содержание элементарного железа (Fe2+или Fe3+) в данной лекарственной форме препарата (капле, таблетке, драже, флаконе) и объём упаковки.
Выбор препарата железа — прерогатива врача. Врач выбирает препарат в соответствии с финансовыми возможностями пациента или его родителей, переносимостью препарата и собственным опытом применения препарата железа.
Вместе с тем каждый врач должен быть информирован об имеющейся в мировой практике тенденции смены солевых препаратов железа, часто демонстрирующих низкую комплайентность, на препараты нового поколения — гидроксид полимальтозный комплекс трёхвалентного железа (Мальтофер*, Феррум-Лек*).
Таблица 15-4. Список некоторых препаратов железа для приёма внутрь
|
Препарат |
Состав препарата (в одном драже, таблетке, в 1 мл капель или сиропа) |
Форма выпуска |
Содержание элементарного железа |
|
Железа сульфат (актиферрин) |
Сульфат железа 113,85 мг, D.L-серин 129 мг в 1 капсуле |
Капсулы, в блистере 10 капсул, по 2 и 5 блистеров в упаковке |
Fe2+: 34,5 мг в капсуле |
|
Железа сульфат (актиферрин) |
Сульфат железа 47,2 мг, D.L-серин 35,6 мг, глюкоза и фруктоза 151,8 мг, калия сорбат 1 мг в 1 мл капель |
Капли для приёма внутрь, 30 мл во флаконе |
Fe2+: 9,48 мг в 1 мл |
|
Железа сульфат (актиферрин) |
Сульфат железа 171 мг, D.L-серин 129 мг, глюкоза, фруктоза в 5 мл сиропа |
Сироп, 100 мл во флаконе |
Fe2+: 34 мг в 5 мл |
|
Железа (III) гидроксид полимальтозат (мальтофер) |
Гидроксид-полимальтозный комплекс |
Раствор для приёма внутрь, 30 мл во флаконе с капельницей |
Fe3+ 50 мг в 1 мл раствора (20 капель) |
|
Железа (III) гидроксид полимальтозат + фолиевая кислота (мальтофер Фол) |
Гидроксид-полимальтозный комплекс, фолиевая кислота 0,35 мг в 1 таблетке |
Жевательные таблетки, 10 таблеток в блистере, по 3 блистера в упаковке |
Fe3+: 100 мг в 1 таблетке |
|
Железа (III) гидроксид полимальтозат (мальтофер) |
Гидроксид-полимальтозный комплекс |
Жевательные таблетки, в блистере 10 таблеток, по 3 и 50 блистеров в упаковке |
Fe3+: 100 мг в 1 таблетке |
|
Железа (III) гидроксид полимальтозат (мальтофер) |
Гидроксид-полимальтозный комплекс |
Сироп, 150 мл во флаконе |
Fe3+: 10 мг в 1 мл |
|
Железа сульфат + аскорбиновая кислота (Сорбифер Дурулес) |
Сульфат железа 320 мг, аскорбиновая кислота 60 мг |
Таблетки, покрытые оболочкой, по 30 и 50 таблеток во флаконе |
Fe3+: 100 мг в 1 таблетке |
|
Железа сульфат (тардиферон) |
Сульфат железа 256,3 мг, мукопротеоза 80 мг, аскорбиновая кислота 30 мг |
Таблетки, покрытые оболочкой, 10 таблеток в блистере, 3 блистера в упаковке |
Fe2+: 80 мг |
|
Тотема |
В 10 мл раствора: 50 мг глюконата железа, 1,33 мг глюконата марганца, 0,7 мг глюконата меди, глицерол, глюкоза, сахароза, лимонная кислота, цитрат натрия и др. |
Раствор для приёма внутрь, ампулы по 10 мл, по 20 ил. в упаковке |
Fe2+: 5 мг в 1 мл |
Окончание табл. 15-4
|
Железа фумарат + фолиевая кислота (ферретаб комп*) |
Фумарат железа 154 мг, фолиевая кислота 0,5 мг |
Капсулы, 10 капсул в блистере, 3 блистера в упаковке |
Fe2+50 мг в 1 капсуле |
|
Железа сульфат + аскорбиновая кислота (ферроплекс) |
Сульфат железа 50 мг, аскорбиновая кислота 30 мг |
Драже, в упаковке 100 шт. |
Fe2+10 мг в 1 драже |
|
Ферронал* |
Глюконат железа 300 мг в 1 таблетке |
Таблетки, покрытые оболочкой, в блистере 10 таблеток, 1 блистер в упаковке |
Fe2+30 мг в таблетке |
|
Хеферол* |
Фумарат железа 350 мг в 1 капсуле |
Капсулы, во флаконе 30 шт. |
Fe2+115 мг в капсуле |
|
Железа (III) гидроксид полимальтозат (Феррум Лек*) |
Г идроксид-полимальтозный комплекс |
Жевательные таблетки, 10 таблеток в стрипе, 3 стри- па в упаковке |
Fe3+100 мг в 1 таблетке |
|
Железа (III) гидроксид полимальтозат (Феррум Лек*) |
Г идроксид-полимальтозный комплекс |
Сироп, 100 мл во флаконе |
Fe3+10 мг в 1 мл |
|
Ферлатум* |
Протеин сукцинилат железа 800 мг в 15 мл |
Раствор для приёма внутрь, 15 мл во флаконе, 10 флаконов в упаковке |
Fe2+40 мг в 15 мл |
|
Поливитамин + минеральные соли (фенюльс*) |
Сульфат железа 150 мг, аскорбиновая кислота 50 мг, рибофлавин 2 мг, тиамин 2 мг, никотинамид 15 мг, пиридоксина гидрохлорид 1 мг, пантотеновая кислота 2,5 мг |
Капсулы, 10 капсул в блистере, 1 блистер в упаковке |
Fe2+45 мг в 1 капсуле |
Недостатки применения солевых препаратов железа при лечении больных ЖДА:
• опасность передозировки, вплоть до отравлений, вследствие негибкого дозирования, пассивного, неконтролируемого всасывания;
• выраженный металлический вкус и окрашивание эмали зубов и дёсен, иногда стойкое;
• взаимодействие с пищей и другими препаратами;
• частый отказ пациентов от лечения (30-35% больных, начавших лечение). Врачи обязаны предупреждать пациентов или их родителей о возможных
отравлениях солевыми препаратами железа. Как показано М.К. Соболевой и О.В. Кольцовым (2002), проанализировавшими все случаи отравлений лекарствами в Новосибирске за 15 лет, отравления препаратами железа составляют всего 1,6% всех случаев отравлений у детей, но в 41,2% случаев заканчиваются летальным исходом.
Свойства и преимущества препаратов на основе гидроксид-полимальтозного комплекса:
• высокая эффективность;
• высокая безопасность: нет риска передозировки, интоксикации и отравления;
• отсутствие потемнения зубов и дёсен;
• приятный вкус, нравится детям;
• отличная переносимость, определяющая регулярность лечения;
• отсутствие взаимодействия с медикаментами и продуктами питания;
• антиоксидантные свойства;
• существование лекарственных форм для всех возрастных групп (капли, сироп, жевательные таблетки, разовые ампулы, препарат железа с фолиевой кислотой для беременных).
Парентеральные (внутримышечные, внутривенные) препараты железа показаны:
• при тяжёлой форме ЖДА (около 3% больных);
• при непереносимости препаратов железа для приёма внутрь;
• при язвенной болезни или операциях на ЖКТ, даже в анамнезе;
• при необходимости быстрого насыщения организма железом.
Расчёт доз
Дозу препарата рассчитывают конкретному больному с учётом:
• степени анемического состояния (I, II, III степень);
• массы тела больного;
• терапевтического плана лечения ЖДА, используемого в данном лечебном учреждении.
Правильный расчёт дозы препарата железа — чрезвычайно важный принцип лечения. Складывается впечатление, что большинство случаев неэффективного лечения препаратами железа связано с неадекватной (заниженной) дозировкой препаратов. Расчёт дозы препаратов железа важен в педиатрической практике, когда врач имеет дело и с новорождёнными детьми, и с подростками, масса тела которых соответствует массе тела взрослого. Используют терапевтический план (табл. 15-5), апробированный у детей, подростков и взрослых.
Таблица 15-5. Терапевтический план лечения железодефицитной анемии в зависимости от степени тяжести
|
Степень тяжести анемии (концентрация Нb, г/л) |
Длительность лечения, мес |
|||
|
1 |
3 |
4 |
6 |
|
|
Доза препарата железа, мг/кг в сут |
||||
|
Лёгкая (110-90) |
5 |
3 |
- |
|
|
Средняя (90-70) |
5-7 |
3-5 |
3 |
- |
|
Тяжёлая (<70) |
8 |
5 |
3 |
|
Длительность курса лечения
Критерием излечения от ЖДА считают преодоление тканевой сидеропении (а не достижение нормального уровня НЫ), что может быть зафиксировано по нормализации уровня СФ. Как показал клинический опыт, для этого требуется не менее 3-6 мес в зависимости от тяжести анемии. Неэффективное лечение препаратами железа и так называемые рецидивы заболевания могут быть связаны с прекращением лечения препаратами железа по достижении нормального уровня Нb.
Контроль эффективности лечения
Эффективность лечения препаратами железа оценивают по нескольким показателям:
• ретикулоцитарной реакции на 7-10-й день от начала лечения препаратами железа;
• началу повышения концентрации Нb после 4 нед лечения препаратами железа (возможно использование критериев ответа на лечение препаратами железа, рекомендуемых американскими специалистами: повышение концентрации Нb на 10 г/л и повышение Ht на 3% по отношению к исходному уровню);
• исчезновению клинических проявлений заболевания через 1-2 мес лечения;
• преодолению тканевой сидеропении, определяемому по уровню СФ, через 3-6 мес от начала лечения (в зависимости от степени тяжести анемии).
Оценка первых 3 показателей особенно важна в случаях, когда врач не имеет возможности провести наиболее информативные лабораторные исследования, подтверждающие ДЖ в организме (MCV, МСНС, МСН, RDW, СЖ, ОЖСС, насыщение трансферрина железом, СФ).
Заместительную терапию эритроцитарной массой следует проводить по строгим показаниям. В настоящее время значительно повышены требования к определению показаний к переливанию компонентов крови у конкретного больного. Врач, назначающий трансфузию, обязан учесть эффект и возможный вред предстоящей гемотрансфузии. С гемотрансфузиями связаны риск трансмиссии различных инфекций (гепатиты, СПИД), образования нерегулярных антител, супрессии собственного гемопоэза - их следует рассматривать как трансплантацию клеток, так как клетки получены от аллогенного донора. Принципиально важно информирование пациента или его родителей (опекунов) о состоянии больного, необходимости трансфузии и связанном с ней риске. Иногда гемотрансфузии невозможны по религиозным соображениям (Свидетели Иеговы). Решение о проведении трансфузии (например, эритроцитарной массы) может принять врач, находящийся в данный момент у постели больного, с учётом:
• характера заболевания;
• тяжести анемии;
• угрозы дальнейшего снижения концентрации Нb;
• переносимости анемии больным;
• стабильности гемодинамических показателей.
Просьба врачей назвать показатели концентрации НЬ, при которых необходимо переливание эритроцитарной массы, — распространённая ошибка, поскольку такой подход не учитывает вышеуказанных показателей. Мнение об отсутствии показаний для переливания эритроцитарной массы при ЖДА, как правило, обосновано. Даже тяжёлые ЖДА можно успешно лечить с помощью препаратов железа для приёма внутрь, внутримышечных или внутривенных препаратов железа.
ЖЕЛЕЗОДЕФИЦИТНЫЕ СОСТОЯНИЯ У ДЕТЕЙ ПЕРВОГО ГОДА ЖИЗНИ И РАННЕГО ВОЗРАСТА
Представление о том, что снижение концентрации Нb у беременной не оказывает влияния на развитие плода, ошибочно. ДЖ у плода приводит к необратимым нарушениям:
• роста массы мозга;
• процесса миелинизации и проведения нервных импульсов через синапсы.
Эти изменения необратимы, их не удаётся корректировать препаратами железа,
назначаемыми в первые месяцы жизни ребёнка. В последующем у ребёнка отмечают задержку психического и моторного развития, нарушение когнитивных функций. Американскими исследователями показано, что даже спустя 5 лет после ЖДА, перенесённой в возрасте 12-23 мес, у ребёнка отмечают задержку умственного и моторного развития, а также трудности с обучением.
Наиболее интенсивный рост отмечается у детей до года и у подростков в пубертатном периоде. Педиатрам известно, что в возрасте 3 мес у многих детей
выявляется сниженный уровень Нb (105-115 г/л). Это явление также было зарегистрировано американскими врачами и послужило основанием для разработки соответствующих рекомендаций. Для детей в возрасте 3 мес была установлена нижняя граница нормы концентрации Нb, соответствующая 95 г/л, поскольку это транзиторное снижение уровня Нb выражено у большинства детей в популяции. Снижение концентрации Нb у большинства детей в 3 мес связано с переходом эри- троидных клеток с синтеза фетального Нb (Hb F) на Нb А2 представляет «физиологическую анемию» и лечения не требует. Концентрацию НЬ следует определять в 6 мес: в этом возрасте её значения соответствуют норме (110 г/л и более).
Если ребёнок находится на грудном вскармливании и не относится к какой- либо группе риска (недоношенность, из многоплодной беременности, рождённый с низкой массой тела), продолжается грудное вскармливание и наблюдение ребёнка. Назначение препаратов железа в профилактических дозах, обычно составляющих 50% лечебной дозы, показано детям из указанных групп риска развития ЖДА.
Постоянный контроль содержания Нb следует осуществлять до 18 мес:
• у детей с низкой массой тела при рождении;
• у недоношенных детей;
• у детей, не получающих молочных смесей, содержащих железо.
С 6-го до 18-го мес уровень Нb следует контролировать, если ребёнок:
• получает коровье молоко до 12 мес;
• на грудном вскармливании после 6 мес получает недостаточное количество железа с прикормом;
• болен (хронические воспалительные заболевания, диетические ограничения, обильная кровопотеря из-за травмы, приём препаратов, нарушающих всасывание железа).
ЖЕЛЕЗОДЕФИЦИТНАЯ АНЕМИЯ У ПОДРОСТКОВ
Подростки, особенно девушки 12-18 лет, нуждаются в скрининге содержания Нb. Целесообразно ежегодное определение уровня Нb у девушек и женщин, имеющих обильные кровопотери при менструации или иной природы, низкое потребление железа с пищей, ЖДА в анамнезе. Не относящиеся к этим группам риска небеременные не нуждаются в частом контроле содержания Нb и могут обследоваться раз в 5 лет, если употребляют пищу, богатую железом и усиливающую его всасывание. Юноши также нуждаются в контроле уровня Нb, если интенсивно занимаются тяжёлыми видами спорта (анемия атлетов). При выявлении ЖДА проводят её лечение.
Проведение профилактических прививок у детей с ЖДС не противопоказано, не требует нормализации уровня Нb, так как количество иммунокомпетентных клеток достаточно.
Россия может и должна опираться на опыт борьбы с ЖДС, полученный в других странах. Наиболее чётко меры профилактики ЖДС сформулированы в национальных «Рекомендациях по профилактике и лечению дефицита железа в США» (1998): первичная профилактика подразумевает правильное полноценное питание, вторичная — активное выявление латентного дефицита железа и ЖДА при диспансеризации, медицинских осмотрах и посещении врача.
Мегалобластные анемии
Межобластные анемии — группа заболеваний, характеризующихся присутствием мегалобластов в костном мозге и макроцитов в периферической крови.
Более чем в 95% случаев мегалобластная анемия развивается в результате дефицита фолатов и витамина В12 или врождённой аномалии их метаболизма.
ЭТИОЛОГИЯ
Выделяют следующие причины развития мегалобластной анемии.
Дефицит витамина В12:
• алиментарный дефицит (содержание в рационе питания витамина В12 <2 мг/ сут; дефицит витамина В12 у матери, приводящий к сниженному содержанию витамина В,в грудном молоке);
• нарушения абсорбции витамина В12;
О недостаточность внутреннего фактора (фактора Кастла):
- пернициозная анемия;
- хирургическое вмешательство на желудке:
- тотальная гастрэктомия;
- частичная гастрэктомия;
- желудочный шунт;
- действие едких веществ;
❖ функциональная аномалия внутреннего фактора;
❖ биологическая конкуренция:
- избыточный рост бактерий в тонкой кишке;
- анастамозы и фистулы;
- слепые петли и карманы;
- стриктуры;
- склеродермия;
- ахлоргидрия;
- гельминты (Diphylobothrium latum);
о- нарушения всасывания в подвздошной кишке:
- семейное избирательное нарушение всасывания витамина В12 (синдром Имерслунд-Гресбека);
- лекарственно индуцированное нарушение всасывания витамина В12;
- хронические заболевания поджелудочной железы;
- синдром Золлингера-Эллисона;
- гемодиализ;
- заболевания, поражающие подвздошную кишку:
- резекция и шунт подвздошной кишки;
- локальный энтерит;
- целиакия;
• нарушения транспорта витамина В12:
❖ наследственный дефицит транскобаламина II;
❖ транзиторный дефицит транскобаламина II;
❖ частичный дефицит транскобаламина I;
• нарушения метаболизма витамина В12:
❖ наследственные:
- дефицит аденозилкобаламина;
- дефицит метилмалонил-КоА-мутазы (mut, mut);
- комбинированный дефицит метилкобаламина и аденозилкобаламина;
- дефицит метилкобаламина;
❖ приобретённые:
- заболевания печени;
- белковая недостаточность (квашиоркор, маразм);
- лекарственно обусловленные (например, аминосалициловая кислота, колхицин, неомицин, этанол, оральные контрацептивы, метформин).
Дефицит фолатов:
• алиментарный дефицит;
• повышенная потребность:
❖ алкоголизм и цирроз печени;
❖ беременность;
❖ новорождённые;
• заболевания, связанные с повышением клеточной пролиферации;
• врождённое нарушение всасывания фолиевой кислоты;
• лекарственно индуцированное нарушение всасывания фолиевой кислоты;
• обширная резекция кишечника, резекция тощей кишки.
Комбинированный дефицит фолиевой кислоты и витамина В12:
• тропическая спру;
• глютен-зависимая энтеропатия.
Врождённые нарушения синтеза ДНК:
• оротовая ацидурия;
• синдром Леша-Нихана ;
• тиамин-зависимая межобластная анемия;
• дефицит ферментов, необходимых для метаболизма фолиевой кислоты:
❖ N5-метил-тетрагидрофолат-трансферазы;
❖ формиминотрансферазы;
❖ дигидрофолат-редуктазы;
• дефицит транскобаламина II;
• аномальный транскобаламин II;
• гомоцистинурия и метилмалоновая ацидурия.
Лекарственно и токсин-индуцированные нарушения синтеза ДНК:
• антагонисты фолатов (метотрексат);
• аналоги пурина (меркаптопурин, азатиоприн, тиогуанин);
• аналоги пиримидина (флуороурацил, 6-азауридин);
• ингибиторы рибонуклеотид-редуктазы (цитозин арабинозид, гидроксикарба- мид);
• алкилирующие агенты (циклофосфамид);
• оксид азота;
• мышьяк;
• хлорэтан.
Кроме того, мегалобластная анемия может быть вызвана эритролейкемией.
Дефицит витамина В12
Витамин В12 (кобаламин — Сbl) поступает в организм в основном с продуктами животного происхождения (такими как мясо, молоко) и усваивается путём абсорбции. Абсорбция витамина В12 — многоэтапный процесс, включающий:
• протеолитическое высвобождение кобаламина из белков;
• присоединение кобаламина к белку желудочного секрета (внутренний фактор — IF, фактор Кастла);
• узнавание комплекса «IF-кобаламин» рецепторами слизистой оболочки подвздошной кишки;
• транспорт через илиальные энтероциты в присутствии ионов кальция;
• высвобождение в циркуляцию системы воротной вены в комплексе с транс- кобаламином II (ТС II) — белком сыворотки крови.
Обычно дефицит витамина В12 у детей раннего возраста вызван его недостаточным поступлением с пищей в организм матери.
Наиболее частое нарушение абсорбции витамина В12 — пернициозная анемия. Это хроническое заболевание, развивающееся вследствие нарушения поступления кобаламина вследствие недостаточности IF в желудочном секрете. Недостаточное содержание IF в желудочном секрете может быть вызвано врождённым дефицитом этого фактора или приобретёнными причинами, в том числе иммунными (выработкой аутоантител против IF и париетальных клеток слизистой оболочки желудка).
Для высвобождения кобаламина из белкового комплекса, в форме которого соединение поступает с пищей, необходимы кислая реакция среды и пепсиновая активность желудочного сока. Именно поэтому пернициозная анемия развивается при некоторых заболеваниях желудка (атрофическом гастрите, частичной гастрэктомии).
При отсутствии или повреждении IF поступление кобаламина в энтероци- ты становится невозможным, что приводит к развитию пернициозной анемии. Недостаточность IF может быть как врождённой, так и приобретённой.
Нарушение метаболизма витамина В12 во многих случаях развивается при недостаточном белковом питании (квашиоркор), заболеваниях печени. Некоторые лекарственные средства влияют на абсорбцию и метаболизм витамина В12.
Дефицит фолиевой кислоты
Дефицит фолиевой кислоты может быть как врождённым, так и приобретённым; последний встречается чаще.
Причины дефицита фолиевой кислоты.
• Неадекватное поступление вследствие:
❖ пристрастий в питании, низкого экономического уровня;
❖ способов приготовления пищи (длительное кипячение приводит к потере 40% фолатов);
❖ вскармливания козьим молоком (1л содержит 6 мкг фолатов);
❖ нарушения питания (квашиоркор, маразм);
❖ специальных диет (при фенилкетонурии, болезни кленового сиропа);
❖ недоношенности;
❖ состояния после трансплантации костного мозга (специальная обработка пищи).
• Нарушения абсорбции:
❖ врождённая изолированная мальабсорбция фолатов;
❖ приобретённая:
- идиопатическая стеаторея;
- тропическая спру;
- полная или частичная гастрэктомия;
- множественные дивертикулы тонкой кишки;
- резекция тощей кишки;
- воспаление подвздошной кишки;
- болезнь Уиппла;
- лимфома кишечника;
- лекарственные препараты: антибиотики широкого спектра действия, дифенилгидантоин (Дилантин), примидон, барбитураты, контрацептивы для приёма внутрь, циклосерин, метформин, этанол, пищевые аминокислоты (глицин, метионин);
- состояние после трансплантации костного мозга (тотальное облучение, лекарственные препараты, поражение кишечника).
• Повышенная потребность:
❖ усиленный рост (недоношенность, беременность);
❖ хронический гемолиз, особенно в сочетании с неэффективным эритропоэзом;
❖ дизэритропоэтические анемии;
❖ злокачественные заболевания (лимфома, лейкоз);
❖ гиперметаболические состояния (например, инфекции, гипертиреоидизм);
❖ обширные поражения кожи (лишаеподобный дерматит, эксфолиативный дерматит);
❖ цирроз печени;
❖ состояние после трансплантации костного мозга (регенерация костного мозга и эпителиальных клеток).
• Нарушения метаболизма фолиевой кислоты:
врождённые:
- дефицит метилен-тетрагидрофолат-редуктазы;
- недостаточность глутамат-формиминотрансферазы;
- функциональная недостаточность 5-метилтетрагидрофолат-гомоцистеин- метилтрансферазы вследствие патологии СblЕ и CblG;
- недостаточность дигидрофолат-редуктазы;
- дефицит метил-тетрагидрофолат-циклогидролазы;
- первичная недостаточность 5-метилтетрагидрофолат-гомоцистеин- метилтрансферазы;
- приобретённые:
- лекарственные препараты: антагонисты фолатов (ингибиторы дигидрофолат-редуктазы): метотрексат, пириметамин, триметоприм, пентамидин®;
- дефицит витамина В12;
- алкоголизм;
- патология печени.
• Повышенная экскреция:
❖ регулярный диализ:
❖ дефицит витамина В12;
❖ заболевания печени;
❖ заболевания сердца.
Алиментарный дефицит фолатов занимает второе место в мире по распространённости среди дефицитных состояний (после алиментарного ДЖ) и развивается в результате недостаточного питания и голодания. Частота заболеваний у женщин выше, чем у мужчин. Запасы фолатов истощаются в течение 3 мес при повышенной потребности в них (во время беременности, в период лактации). При недостаточном содержании фолатов в организме плода его нервная система развивается неправильно. Именно поэтому до зачатия и во время беременности женщинам назначают фолиевую кислоту с профилактической целью. Недостаточное поступление фолиевой кислоты в период беременности приводит к преждевременным родам и рождению детей с низкой массой тела. При рождении клинические проявления дефицита фолатов наблюдаются редко. Быстрый рост в первые несколько недель жизни ребёнка сопровождается повышенной потребностью в фолиевой кислоте, поэтому в этот период с профилактической целью рекомендуют назначать препарат по 0,05-0,2 мг в сут.
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ НЕДОСТАТОЧНОСТИ КОБАЛАМИНА И ФОЛАТОВ
Начальные проявления (могут наблюдаться в течение нескольких месяцев до появления развёрнутой клинической картины):
• мегалобластная анемия;
• парестезии;
• болезненность языка или всей ротовой полости;
• красный гладкий («лакированный») язык;
• потеря массы тела (как результат анорексии);
• трудности при ходьбе и выполнении тонких движений руками;
• усталость;
• летаргия.
Развёрнутые клинические проявления:
• нарушения пигментации:
❖ нежно-жёлтая окраска кожи (сочетание бледности с лёгкой иктеричностью);
появление очагов гиперпигментации и витилиго;
❖ обесцвечивание волос;
• фебрильная лихорадка (часто встречается);
• поражения ЖКТ:
• глоссит;
❖ потеря аппетита (до анорексии), тошнота, рвота;
❖ стул кашицей несколько раз в день или диарея, метеоризм;
❖ иногда псевдоопухоль брюшной полости за счёт гипертрофии мышц пилорического сфинктера;
❖ эпизодические боли в животе различной интенсивности;
• поражения нервной системы в виде периферической нейропатии (за счёт дегенеративных процессов в задних и латеральных отделах спинного мозга, а также в периферических нервах):
❖ апатия, слабость;
❖ раздражительность;
❖ задержка психомоторного развития и потеря навыков, особенно двигательных, у детей раннего возраста;
❖ наличие непроизвольных движений;
❖ мышечная гипотония, отсутствие рефлексов;
❖ парестезии конечностей, отсутствие чувствительности;
❖ потеря ориентации на фоне нарушения походки;
❖ положительная проба Ромберга;
❖ спастический парез с повышением коленных и голеностопных рефлексов;
❖ появление рефлекса Бабинского.
ДИАГНОСТИКА
При сборе анамнеза пациента обращают внимание на:
• длительный приём антибиотиков и антиконвульсантов;
• тип диеты/питания;
• наличие и длительность диареи;
• хирургические вмешательства на ЖКТ.
Общий анализ крови выявляет:
• анемию;
• повышение эритроцитарных индексов (MCV — может быть до 110-140 фл, RDW);
• макроцитоз эритроцитов;
• много макроовалоцитов;
• выраженный анизопойкилоцитоз эритроцитов;
• наличие телец Жолли и колец Кебота;
• лейкопению (до 1,5х109/л);
• гиперсегментацию ядер нейтрофилов (5 и более сегментов);
• тромбоцитопению (до 50х109/л).
При исследовании костного мозга обнаруживают признаки мегалобластного типа кроветворения:
• клетки крупные;
• ядра зернистые, исчерченные;
• цитоплазма клеток более зрелая по сравнению с ядром;
• диссоциация ядра и цитоплазмы сильнее выражена в более зрелых клетках;
• встречаются клетки, содержащие ядра со слабоконденсированным хроматином;
• множественные, иногда патологические митозы;
• остатки ядер, тельца Жолли;
• клетки, содержащие 2 или 3 ядра;
• качественные нарушения эритропоэза;
• огромные (гигантские) метамиелоциты с подковообразными ядрами;
• гиперсегментацию нейтрофилов;
• многоядерные мегакариоциты.
Результаты анализа мочи выявляют стойкую протеинурию (признак специфического нарушения всасывания витамина В12 в подвздошной кишке).
Определяют:
• уровень витамина В12 в сыворотке крови: нормальные значения - 200- 300 пг/мл;
• уровень фолатов в сыворотке крови: нормальное значение — более 5-6 нг/мл (низкое — менее 3 нг/мл, пограничное — 3-5 нг/мл);
• уровень фолатов в эритроцитах: нормальное значение — 74-640 нг/мл;
• уровень экскреции оротовой кислоты для диагностики оротурии.
Деоксиуридиновый тест проводят для дифференциации между дефицитом витамина В12 и фолиевой кислоты.
Тест Шиллинга проводят для определения активности IF и абсорбции витамина В12 в кишечнике.
Тест Шиллинга с использованием коммерческого IF проводят в случае нарушенного простого теста Шиллинга для дифференциации между патологией IF и специфическим нарушением всасывания витамина В12 в подвздошной кишке (синдром Имерслунд-Гресбека) или дефицитом транскобаламина II. В случае наслоения бактериальной инфекции тест следует повторить после курса антибактериальной терапии (после лечения тетрациклином тест обычно приходит в норму).
Проводят исследование желудочной кислотности (исходной и после стимуляции гистамином), содержания IF в желудочном соке (в том числе после добавления соляной кислоты для выявления в желудочном соке антител к IF), биопсию слизистой оболочки желудка.
Определяют антитела к IF и париетальным клеткам в сыворотке крови.
Кроме того, определяют уровень голотранскобаламина II в сыворотке крови: при дефиците витамина В12 концентрация голотранскобаламина II (кобаламина, связанного с транскобаламином II) существенно ниже нормальных значений, что предшествует снижению уровня общего кобаламина сыворотки крови.
В сыворотке крови и в моче определяют концентрацию метилмалоновой кислоты и гомоцистеина: при дефиците фолатов содержание метилмалоновой кислоты в пределах нормы, гомоцистеина — повышено.
Для диагностики врождённой метилмалоновой ацидурии возможно определение метилмалоната в амниотической жидкости или в моче беременной.
Проводят тесты на мальабсорбцию.
ЛЕЧЕНИЕ
Лечение дефицита витамина В12
Профилактику проводят в случае гастрэкомии и резекции подвздошной кишки.
Начальная суточная доза витамина В12 составляет 0,25-1,0 мг (250-1000 мкг) в течение 7-14 дней. В качестве альтернативной схемы (при способности организма надолго запасать витамин) используют внутримышечное введение препарата в дозе 2-10 мг (2000-10 000 мкг) ежемесячно. В большинстве случаев терапию проводят пожизненно.
При дефиците транскобаламина II терапевтический ответ достигается введением высоких доз витамина В12, содержание кобаламина в сыворотке крови необходимо поддерживать на достаточно высоком уровне. Адекватный контроль заболевания обеспечивается введением внутримышечно 10 мг (10 000 мкг) витамина В12 2-3 раза в нед.
При метилмалоновой ацидурии и нарушении синтеза коферментов кобаламина витамин В12 назначают в дозе 0,01-0,02 мг (10-20 мкг) в сут, однако для некоторых больных эта доза недостаточна. Возможно введение препарата путём кордоцентеза.
При В12-дефицитной мегалобластной анемии отмечается повышение количества ретикулоцитов на 3-4-й день лечения, максимально — на 6-8-й день, к 20-му дню лечения количество ретикулоцитов нормализуется. Содержание ретикулоцитов обратно пропорционально степени выраженности анемии. В костном мозге мегалоцитоз начинает исчезать через 6 ч после введения витамина В12 и через 72 ч после начала лечения полностью отсутствует. Снижение выраженности неврологических симптомов отмечают через 48 ч, отставание в психомоторном развитии прекращается через несколько месяцев. Часто у больных отмечают остаточные неврологические изменения.
Необходимо помнить, что назначение фолиевой кислоты больным с мегалобластной анемией вследствие дефицита витамина В12 приводит к обратному развитию только гематологической симптоматики, а неврологическая в большинстве случаев прогрессирует или остаётся неизменной, поэтому её применение противопоказано.
Профилактика дефицита фолиевой кислоты
Профилактика дефицита фолиевой кислоты состоит в коррекции питания (устранении симптомов мальабсорбции и причин алиментарного дефицита фолатов). Назначают приём препаратов фолиевой кислоты коротким курсом. Пожизненно фолиевую кислоту в дозе 1-2 мг в сут назначают:
• при хроническом гемолизе (например, талассемии):
• при неэффективности аглиадиновой диеты;
• при синдроме мальабсорбции.
Лечение дефицита фолиевой кислоты
Оптимальный результат лечения достигается при назначении фолиевой кислоты в дозе 100-200 мкг в сут. Одна таблетка содержит 0,3-1,0 мг фолиевой кислоты, раствор для инъекций — 1 мг/мл. Длительность терапии составляет несколько месяцев, пока не сформируется новая популяция эритроцитов.
При терапии наследственной недостаточности дигидрофолат-редуктазы лечение проводят не фолиевой кислотой, а N-5-формилтетрагидрофолиевой кислотой.
Обратное развитие симптомов дефицита фолиевой кислоты происходит довольно быстро. В течение 24-48 ч после приёма препарата снижается уровень СЖ (обычно до низких значений); уровень ретикулоцитов повышается к 2-4-му дню лечения, достигая пика на 4-7-е сут. Одновременно с повышением числа ретикулоцитов увеличивается содержание лейкоцитов и тромбоцитов. Мегалобластные изменения в костном мозге исчезают в течение 24-48 ч от начала терапии, однако крупные миелоциты и метамиелоциты могут сохраняться в течение нескольких месяцев. Улучшение аппетита и общего состояния больного отмечают через 1-2 дня. Уровень Нb нормализуется на 2-6-й нед лечения.
НАСЛЕДСТВЕННЫЕ ГЕМОЛИТИЧЕСКИЕ АНЕМИИ
Главный признак гемолитической анемии - уменьшение продолжительности жизни эритроцитов (<120 дней). Разрушение эритроцитов может быть обусловлено как внутриклеточными (патологией мембраны, ферментов, гемоглобина), так и внеклеточными причинами. Алгоритм диагностики гемолитической анемии включает оценку совокупности клинических проявлений заболевания, характерных лабораторных изменений, выявление причины возникновения гемолиза.
ПРИЧИНЫ РАЗВИТИЯ ГЕМОЛИТИЧЕСКОЙ АНЕМИИ
ВСЛЕДСТВИЕ АНОМАЛИИ ЭРИТРОЦИТОВ
Дефекты мембраны эритроцитов (мембранопатии):
• первичные мембранопатии со специфическими морфологическими нарушениями (изменениями белкового состава мембраны):
❖ наследственный сфероцитоз;
❖ наследственный овалоцитоз;
❖ наследственный стоматоцитоз:
- со сниженной осмотической резистентностью эритроцитов;
- с повышенной осмотической резистентностью эритроцитов;
- с нормальной осмотической резистентностью эритроцитов;
- с RhO;
❖ врождённая гемолитическая анемия с дегидратацией эритроцитов (повышено содержание Na+, понижено содержание К4, повышена осмотическая резистентность эритроцитов);
• изменение фосфолипидного состава мембраны;
• наследственная недостаточность АТФазы;
• вторичные дефекты мембран эритроцитов — абеталипопротеинемия. Дефекты ферментов эритроцитов (ферментопатии):
• нарушения анаэробного гликолиза — дефицит:
❖ пируваткиназы;
❖ гексокиназы;
❖ глюкозофосфат-изомеразы;
❖ фосфофруктокиназы;
❖ альдолазы;
❖ триозофосфат-изомеразы;
❖ фосфоглицераткиназы;
• нарушения пентозофосфатного шунта:
❖ дефицит Г-6-ФД;
❖ дефицит 6-фосфоглюконатдегидрогеназы;
❖ дефекты синтеза глутатиона;
- дефицит глутатионредуктазы;
- дефицит глутатионпероксидазы;
- дефицит глутатион-S-трансферазы;
• нарушения метаболизма нуклеотидов:
❖ дефицит пиримидин-5’-нуклеотидазы;
❖ дефицит аденилаткиназы;
❖ избыток аденозин-деаминазы.
Нарушения синтеза гемоглобина:
• нарушения синтеза гема:
❖ врождённая сидеробластная анемия;
❖ врождённая эритропоэтическая порфирия;
• нарушения синтеза глобина:
❖ качественные гемоглобинопатии (аномальные гемоглобины);
❖ количественные гемоглобинопатии (талассемии).
Врождённые дизэритропоэтические анемии:
• тип I;
• тип II;
• тип III;
• тип IV.
ПРИЧИНЫ РАЗВИТИЯ ГЕМОЛИТИЧЕСКОЙ АНЕМИИ,
НЕ СВЯЗАННЫЕ С АНОМАЛИЯМИ ЭРИТРОЦИТОВ
Иммунные причины развития гемолитической анемии:
• изоиммунные:
❖ гемолитическая болезнь новорождённых;
❖ трансфузия несовместимой крови;
• аутоиммунные — IgG; комплемент; смешанные — IgG + комплемент:
❖ идиопатические:
- с тепловыми антителами;
- с холодовыми антителами;
- с антителами Доната-Ландштейнера;
❖ вторичные:
- при инфекционных заболеваниях, вызванных вирусами (Эпстайна-Барр, цитомегаловирусом, гепатита, простого герпеса, кори, краснухи, гриппа типа А, коксаки типа В, ВИЧ) или бактериями (стрептококками, брюшным тифом, Escherichia coli (септицемией), Mycoplasma pneumonia);
- при приёме лекарственных препаратов и действии химических агентов (хинина, хинидина, фенацитина, σ-аминосалициловой кислоты, цефалотина, препаратов пенициллина, тетрациклина, рифампицина, сульфаниламидов, хлорпромазина, пирамидона, дипирона, инсулина свинца);
- при гематологических заболеваниях (лейкозах, лимфомах, лимфопролиферативном синдроме, ИТП — синдроме Эванса, пароксизмальной холодовой гемоглобинурии, пароксизмальной ночной гемоглобинурии);
- при иммунных заболеваниях (СКВ, узелковом периартериите, склеродермии, дерматомиозите, ревматоидном артрите, неспецифическом язвенном колите, агаммаглобулинемии, синдроме Вискотта-Олдрича, дисгаммагло- булинемии, недостаточности IgA, аутоиммунном тиреоидите, гепатите, аутоиммунной гемолитической анемии с положительной пробой Кумбса);
- при опухолях (тератоме яичников, дермоидных опухолях, тимоме, карциноме).
Неиммунные причины развития гемолитической анемии:
• идиопатические;
• вторичные:
❖- инфекции:
- вирусные (инфекционный мононуклеоз, вирусный гепатит);
- бактериальные (стрептококковые, септицемия Е. coli, Clostridium perfrin-gens, Bartonella bacilliformis);
- паразитарные (малярия, гистоплазмоз);
❖ лекарственные препараты и химические агенты:
- фенилгидрозин;
- витамин К;
- нитробензин;
- сульфоны;
- фенацетин®;
- ацетиламид;
- свинец;
❖ гематологические заболевания:
- лейкоз;
- АА;
- мегалобластная анемия;
- пикноцитоз;
❖ микроангиопатическая гемолитическая анемия:
- ТТП;
- гемолитикоуремический синдром;
- хроническая рецидивирующая шистоцитарная гемолитическая анемия;
- ожоги;
- маршевая гемоглобинурия;
- искусственные клапаны сердца;
❖ другие:
- болезнь Вильсона-Коновалова;
- эритропоэтическая пурпура;
- остеопороз;
- гиперспленизм.
Клинические признаки, свидетельствующие о наличии гемолиза:
• бледность кожных покровов и слизистых оболочек;
• желтушность кожных покровов и слизистых оболочек;
• тёмная моча (возможна связь с приёмом медикаментов или контактом с химическими агентами);
• увеличение печени и/или селезёнки.
При сборе данных анамнеза учитывают:
• этническую принадлежность (выявление Hb S у лиц негроидной расы, дефицита глюкозо-6-фосфатдегидрогеназы у лиц еврейской национальности, талассемии у жителей Средиземноморья, Средней Азии, Республик Кавказа);
• возраст [выявление у новорождённых детей с положительным резус-фактором, рождённых от матерей с отрицательным резус-фактором; у детей с группой крови А(ІІ) или В(ІІІ), рождённых от матерей с группой крови 0(1)];
• наличие желчнокаменной болезни у больного и/или его родителей;
• персистирующую анемию с повышенным количеством ретикулоцитов;
• хронические трофические язвы голеней.
Лабораторные показатели характеризуют повышенное разрушение эритроцитов с усилением катаболизма Нb и усиление эритропоэза.
Лабораторные тесты, применяемые для выявления гемолиза
Об усилении катаболизма гемоглобина свидетельствуют:
• уровень билирубина сыворотки крови (общий и непрямой);
• уровень уробилирубина в моче;
• уровень уробилирубина в кале;
• уровень гаптоглобина в плазме крови;
• концентрация свободного НЬ в плазме крови;
• уровень метгемальбумина в плазме крови;
• уровень метгемоглобина в плазме крови;
• содержание гемосидерина в моче;
• содержание Нb в моче;
• содержание дипирролов в моче;
• морфология эритроцитов периферической крови;
• исследование продолжительности жизни эритроцитов (51Сr, дифлуорофосфат - 32DFP).
Об усилении эритропоэза свидетельствуют:
• число ретикулоцитов в периферической крови;
• наличие и число нормобластов в периферической крови;
• исследование костного мозга (гиперплазия эритроидного ростка);
• рентгенография плоских костей («волосковые» изменения).
Скорость катаболизма Нb зависит от типа гемолиза (внутриклеточного или внутрисосудистого). При внутриклеточном гемолизе выявляется повышение уровня билирубина за счёт непрямой фракции, количества уробилина в кале и моче. При внутрисосудистом гемолизе отмечаются:
• повышение уровня свободного Нb плазмы крови;
• гемоглобинурия;
• гемосидеринурия;
• понижение уровня гаптоглобина в плазме крови вплоть до его полного отсутствия;
• повышение уровня метгемальбумина (соединение свободного гема с альбумином) и метгемоглобина плазмы крови.
Причины острой гемоглобинурии:
• трансфузия несовместимой крови;
• действие медикаментов и химических агентов;
• инфекции (Closbidium perfringens, Bartonella bacilliformis, малярийный плазмодий);
• ожоги;
• механические причины (искусственный клапан сердца).
Гемолиз часто вызывают:
• лекарственные средства:
❖ фенилгидразин;
❖ сульфоны;
❖ фенацетин;
❖ ацетиламид в больших дозах;
• химические агенты:
❖ нитробензин;
❖ свинец;
❖ случайная инфузия воды;
• токсины:
❖ укусы змей;
❖ укусы пауков.
Редко вызывают гемолиз:
• при дефиците Г-6-ФД или аномального нестабильного Нb:
❖ сульфаниламиды;
❖ нитрофураны;
❖ противомалярийные препараты;
антипиретики;
❖ витамин К;
❖ употребление в пищу плодов растений семейства бобовых (фавизм);
❖ нафталин;
• при повышенной чувствительности:
❖ хинин;
❖ хинидин;
❖ парааминосалициловая кислота;
❖ фенацитин®.
Причины хронической гемоглобинурии:
• пароксизмальная холодовая гемоглобинурия;
• пароксизмальная ночная гемоглобинурия;
• маршевая гемоглобинурия;
• гемолиз с холодовыми агглютининами.
Эритропоэз усиливается в ответ на снижение уровня Нb, этот процесс характеризуется:
• ретикулоцитозом и появлением нормобластов в периферической крови;
• расширением эритроидного ростка костного мозга;
• экспансией кроветворного костного мозга (деформацией плоских костей скелета);
• укорочением продолжительности жизни эритроцитов (при использовании изотопа 51Cr);
• специфической морфологией эритроцитов.
Для уточнения причины гемолиза необходимо проведение дополнительного обследования.
Необходимые лабораторные исследования для выявления причины гемолиза:
• вызванного внутренними причинами (аномалиями эритроцитов):
❖ мембранопатиями:
- морфология и морфометрия эритроцитов;
- осмотическая резистентность эритроцитов по Идельсону (с инкубацией и без неё);
- пробы на аутогемолиз;
- электрофорез мембранных белков;
- электрофорез мембранных липидов;
- сканирующая электронная микроскопия;
❖ гемоглобинопатиями:
- морфология эритроцитов;
- проба на серповидность;
- электрофорез Нb;
- тест на стабильность гемоглобина;
- сродство Нb к кислороду;
- биосинтез цепей глобина;
- количество фетального Нb;
- окраска на тельца Гейнца;
❖ ферментопатиями:
- качественные тесты на дефицит ферментов;
- тест на аутогемолиз;
- количественное определение активности ферментов;
• вызванного внешними причинами:
❖ проба Кумбса прямая и непрямая;
❖ тест Хема;
тест Доната-Ландштейнера.
Мембранопатии
НАСЛЕДСТВЕННЫЙ СФЕРОЦИТО3
Чаще встречается у жителей Северной Европы, где распространённость заболевания составляет 1 на 5000 населения.
Аутосомно-доминантный тип наследования встречается примерно в 75% случаев заболевания. У членов семьи пациента тяжесть анемии и степень сфероцитоза может варьировать. В 25% случаев семейный анамнез отсутствует. У части пациентов изменения лабораторных показателей минимальны, что предполагает аутосомно-рецессивный тип наследования, остальные случаи — результат спонтанных мутаций.
Первичный дефект при наследственном сфероцитозе — нестабильность мембраны эритроцитов вследствие нарушения функции или недостаточности скелетного белка эритроцитов. Наиболее характерен дефект спектрина и/или анкирина, однако может быть и дефицит других скелетных белков: белка полосы 3, белка полосы 4.2. Обычно (75-90%) встречается дефицит спектрина. Тяжесть заболевания, а также степень сфероцитоза (по оценке осмотической резистентности и морфометрии эритроцитов) зависят от степени дефицита спектрина. У гомозиготных пациентов с уровнем спектрина, составляющим до 30-50% нормального, развивается выраженная гемолитическая анемия, часто трансфузионно зависимая. Недостаточность анкирина наблюдается примерно у 50% детей, родители которых здоровы. Риск развития заболевания у других детей составляет менее 5%.
Вследствие дефицита скелетного белка развиваются следующие нарушения:
• потеря мембраной липидов;
• уменьшение соотношения площади поверхности клетки к её объему (потеря поверхности);
• изменение формы эритроцитов (сфероцитоз);
• ускорение поступления натрия в клетку и его выхода из клетки, что вызывает дегидратацию клетки;
• быстрая утилизация АТФ с усилением процесса гликолиза;
• разрушение незрелых форм эритроцитов;
• секвестрация эритроцитов в системе фагоцитирующих макрофагов селезенки.
Изменения гематологических показателей;
• анемия различной степени тяжести, при апластическом кризе — до 20-30 г/л;
• ретикулоцитоз;
• некоторое снижение MCV;
• повышение МСНС и RDW.
При морфологической оценке эритроцитов периферической крови выявляют микросфероцитоз различной степени выраженности, отмечают повышенную плотность клеток, полихромазию. Содержание микросфероцитов — главный маркёр тяжести заболевания. Повышенная плотность клеток чаще наблюдается при серповидноклеточных состояниях. В случае наследственного сфероцитоза такие клетки отражают не степень тяжести заболевания, а черты фенотипа. Средний диаметр эритроцитов снижен (<7,5 мкм), индекс сфероцитоза снижен (<3,5), индекс овалоцитоза не изменён (0,78-1,0).
Снижение осмотической резистентности эритроцитов — лизирование эритроцитов в более высоких концентрациях раствора натрия хлорида по сравнению с нормальными клетками — как до, так и после инкубации (24 ч при 37 °С). Увеличение МСНС или плотности эритроцитов при нормальной осмотической резистентности эритроцитов свидетельствует о наследственном сфероцитозе.
Наблюдается повышение аутогемолиза через 24 и 48 ч, корригируемое добавлением глюкозы.
Уменьшается продолжительность жизни 51Сr-меченых эритроцитов, характерна их повышенная секвестрация в селезёнке.
В костном мозге обнаруживаются нормобластическая гиперплазия эритроидного ростка, повышение уровня железа.
Изменения в биохимическом анализе крови:
• повышение общего билирубина за счёт непрямой фракции;
• при обтурационной желтухе вследствие желчнокаменной болезни, осложнившей наследственный сфероцитоз, возможно повышение прямой фракции билирубина.
Клиническая картина заболевания
Наблюдаются анемия и желтуха, их тяжесть зависит от степени гемолиза, ретикулоцитоза и способности печени конъюгировать и экскретировать непрямой билирубин. Характерна спленомегалия.
Диагностику заболевания осуществляют по клинической картине, данным семейного анамнеза и перечисленным выше специфическим гематологическим показателям.
Осложнения
Гемолитический криз — резкое усиление процессов гемолиза, нередко на фоне инфекции.
Эритробластопенический (апластический) криз — арест созревания клеток эритроидного ряда — часто связан с межобластными изменениями. Обычно про
воцируется парвовирусной инфекцией В19, так как парвовирус В19 поражает развивающиеся нормобласты, вызывая транзиторное прекращение их продукции.
Дефицит фолатов за счёт ускоренного оборота эритроцитов может привести к развитию ярко выраженной мегалобластной анемии.
Желчнокаменная болезнь встречается примерно у половины нелеченых больных, с возрастом вероятность её развития повышается.
Вторичная перегрузка железом встречается редко.
Лечение
Назначают фолиевую кислоту по 1 мг в сут в период гемолиза.
При тяжёлом гемолитическом или апластическом кризе (Нb <60 г/л) проводят заместительную терапию эритроцитарной массой, очищенной от лейкоцитов.
Спленэктомию применяют в плановом порядке. Перед проведением оперативного лечения необходимо вакцинировать больного против менингококковой и пневмококковой инфекции, а также гемофильной инфекции типа В.
При желчнокаменной болезни применяют холецистэктомию.
Диспансерное наблюдение
Рекомендуют осмотр гематолога, общий анализ крови с подсчётом ретикулоцитов (раз в месяц при среднетяжёлой и тяжёлой формах, раз в квартал — при лёгкой и минимальной формах).
Биохимический анализ крови (определение содержания общего и непрямого билирубина, активности АЛТ, ACT, щелочной фосфатазы, ЛДГ, концентрации СЖ, ОЖСС, насыщение трансферрина железом, СФ) проводят раз в квартал при среднетяжёлой и тяжёлой формах, раз в 6 мес — при лёгкой и минимальной формах.
УЗИ органов брюшной полости рекомендуют раз в 6-12 мес.
Ферментопатии эритроцитов
Различают 2 основных биохимических пути энергетического обмена в эритроцитах: гликолитический путь Эмбдена-Мейергофа (анаэробный) и пентозофосфатный гликолиз. При дефектах любого из ферментов, участвующих в этих процессах, развивается гемолиз.
ДЕФИЦИТ ПИРУВАТКИНАЗЫ
Пируваткиназа — один из основных ферментов гликолитического пути. Пируваткиназа катализирует превращение фосфоэнолпирувата в пируват и, таким образом, участвует в гликолитической реакции образования АТФ (аденозинтрифосфата). Фермент аллостерически активируется фруктозо-1,6-дифосфатом (Ф-1,6-ДФ) и ингибируется образующейся АТФ. При дефиците пируваткиназы в эритроцитах накапливается 2,3-дифосфоглицерат и другие продукты гликолиза. Концентрация АТФ, пирувата и лактата в эритроцитах снижена. Парадоксально то, что концентрация аденозинмонофосфата (АМФ) и АДФ в эритроцитах также снижена в основном за счёт зависимости АТФ от фосфорибозилпирофосфат-синтетазы и других ферментов, вовлечённых в синтез адениновых нуклеотидов. Дефицит АТФ также влияет на синтез никотинамидадениндинуклеотида (НАД). Поскольку уровень гликолиза ограничен доступностью (количеством) НАД, недостаточный синтез НАД способствует дальнейшему уменьшению образования АТФ и провоцирует гемолиз эритроцитов. Заболевание наследуется по аутосомно-рецессивному типу.
Гематологические показатели
В общем анализе крови обнаруживают признаки гемолитической несфероцитарной анемии:
• концентрацию Нb — 60-120 г/л;
• гематокрит — 17-37%;
• нормохромию;
• нормоцитоз (у детей до года и при высоком ретикулоцитозе возможен макроцитоз);
• ретикулоциты 2,5-15%, после спленэктомии — до 70%;
• морфологические признаки:
❖ полихромазию эритроцитов;
❖ анизоцитоз;
❖ пойкилоцитоз;
❖ возможно наличие нормобластов.
Осмотическая резистентность эритроцитов до инкубации не изменена, после инкубации снижена, корригируется добавлением АТФ.
Аутогемолиз значительно повышен, корригируется добавлением АТФ, но не глюкозы.
Активность пируваткиназы эритроцитов снижена до 5-20% нормальной, содержание 2,3-дифосфоглицерата и других промежуточных метаболитов гликолиза повышено в 2-3 раза; за счёт повышения содержания 2,3-дифосфоглицерата кривая диссоциации кислорода сдвинута вправо (снижено сродство Нb к кислороду).
Скрининговый тест базируется на флюоресценции НАДН в ультрафиолетовом свете: к тестируемой крови добавляют фосфоэнолпируват, НАДН и лактатдегидрогеназу, наносят на фильтровальную бумагу и исследуют в ультрафиолетовом свете. При дефиците пируваткиназы пирувата не образуется, и НАДН не используется, вследствие этого флюоресценция сохраняется в течение 45-60 мин. В норме флюоресценция исчезает через 15 мин.
Клиническая картина
Тяжесть состояния варьирует, может отмечаться анемия тяжёлой степени, не индуцированная приёмом лекарственных средств. Желтуха обычно развивается с рождения. Почти всегда присутствует спленомегалия. С возрастом развиваются желчнокаменная болезнь, вторичная перегрузка железом и изменение костей скелета (вследствие частых трансфузий эритроцитарной массы). Апластические кризы провоцируются парвовирусной В19-инфекцией.
Лечение
Фолиевая кислота по 0,001 г/сут ежедневно.
Заместительная терапия эритроцитарной массой для поддержания уровня Нb более 70 г/л.
Спленэктомию применяют только при повышении потребности в трансфузиях эритроцитарной массы свыше 200-220 мл/кг в год (при Ht эритроцитарной массы 75%), спленомегалии, сопровождающейся болями в левом подреберье и/или угрозой разрыва селезёнки, а также при явлениях гиперспленизма. Перед проведением оперативного лечения необходимо вакцинировать пациента против менингокок- ковой, пневмококковой и гемофильной инфекции типа В.
Нежелательно использовать салицилаты, так как в условиях дефицита пируваткиназы салицилаты провоцируют нарушение окислительного фосфорилирования в митохондриях.
ДЕФИЦИТ ГЛЮКОЗО-6-ФОСФАТДЕГИДРОГЕНАЗЫ
Г-6-ФД — первый фермент пентозофосфатного гликолиза. Основная функция фермента заключается в восстановлении НАДФ до НАДФН, необходимого для перехода окисленного глутатиона (GSSG) в восстановленную форму. Восстановленный глутатион (GSH) требуется для связывания активных форм кислорода (перекисей). Пентозофосфатный гликолиз обеспечивает клетку энергией.
Недостаточность активности фермента снижает энергетические запасы клетки и приводит к развитию гемолиза, тяжесть которого зависит от количества и варианта Г-6-ФД. В зависимости от тяжести дефицита выделяют 3 класса вариантов Г-6- ФД. Дефицит Г-6-ФД сцеплен с Х-хромосомой, наследуется рецессивно. Больные мужского пола всегда гемизиготны, женского — гомозиготны.
Клиническая картина
Тяжесть состояния варьирует, может развиваться анемия тяжёлой степени, не индуцированная лекарственными препаратами и/или химическими агентами.
Желтуха обычно развивается с рождения. Почти всегда присутствует спленомегалия. В отдалённые сроки часто образуются камни в жёлчном пузыре, развивается вторичная перегрузка железом, характерны изменения костей.
Наблюдаются апластические кризы, провоцируемые парвовирусной инфекцией В19.
Лечение
Необходимо исключить приём лекарственных препаратов, провоцирующих гемолиз. Рекомендуют приём фолиевой кислоты.
При снижении концентрации Нb менее 60 г/л проводят заместительную терапию эритроцитарной массой (требования к качеству и расчёт объёма эритроцитарной массы представлены ниже).
Спленэктомию применяют только при развитии вторичного гиперспленизма, так как операция не приводит к прекращению гемолиза.
Гемоглобинопатии
Гемоглобинопатии — наследственное качественное (аномальные гемоглобины) или количественное (талассемии) нарушение синтеза глобиновых цепей.
КОЛИЧЕСТВЕННЫЕ ГЕМОГЛОБИНОПАТИИ (ТАЛАССЕМИИ)
В зависимости от дефицита той или иной глобиновой цепи, выделяют β- и α-талассемии.
β-Талассемия
β-Талассемия (β-тал) — гетерогенная группа заболеваний, характеризующаяся уменьшением или отсутствием синтеза (3-глобиновых цепей. В зависимости от тяжести состояния выделяют 3 формы (3-тал: большую, промежуточную и малую. Тяжесть клинических проявлений прямо пропорциональна степени дисбаланса глобиновых цепей. В зависимости от степени снижения синтеза [3-глобиновых цепей выделяют:
• β°-талассемию (β°-тал), при которой синтез β-глобиновых цепей полностью отсутствует;
• β+-талассемию (β+-тал), при которой синтез β-глобиновых цепей сохранён. Малая форма β-талассемии
У гетерозиготных пациентов болезнь, как правило, протекает бессимптомно, уровень Нb соответствует нижней границе нормы или слегка снижен. Индексы MCV и МСН снижены до типичного уровня в 60-70 фл (норма — 85-92 фл) и 20-25 пг (норма — 27-32 пг) соответственно. Гематологические характеристики также включают:
• микроцитоз;
• гипохромию;
• анизопойкилоцитоз с мишеневидностью и базофильной пунктацией эритроцитов периферической крови;
• незначительное расширение эритроидного ростка в костном мозге. Увеличение селезёнки развивается редко и обычно выражено незначительно.
Диагностическими тестами считают электрофорез Нb и подсчёт уровня Нb А2,
как правило, вдвое превышающего норму (норма соответствует 2,0-3,5%). Уровень Hb F в половине случаев слегка повышен — не превышает 5%. Биосинтез in vitro цепей глобина выявляет дефицит p-цепей: соотношение α/β составляет
1,5-2,0 (норма — 0,9-1,1). Изредка значения концентрации Hb F, Нb А2, индексов MCV и МСН у гетерозиготных пациентов могут быть нормальными.
Большая форма β-талассемии
Типичные клинико-гематологические черты заболевания появляются на 6-м месяце жизни, после переключения на взрослый тип глобинового синтеза, и проявляются в виде тяжёлой гипохромной микроцитарной гемолитической анемии, требующей регулярных гемотрансфузий для поддержания адекватного уровня Нb. У детей, не получающих заместительной терапии, тяжёлая анемия приводит к высокой ранней смертности в возрасте 3-4 лет при β°-тал или в возрасте 8-12 лет при р -тал. К клиническим проявлениям также относят:
• прогрессирующее увеличение печени и селезёнки;
• типичные изменения плоских костей вследствие экспансии костного мозга;
• патологические переломы трубчатых костей.
Вторичный гиперспленизм, приводящий к тромбоцитопении, лейкопении и ускоренному разрушению перелитых эритроцитов, ещё больше осложняет клиническую картину. Задержка роста и полового развития начинает обращать на себя внимание к 10 годам. Течение заболевания у неадекватно леченных больных осложняется перегрузкой железа в результате трансфузий и повышением всасываемости железа в кишечнике. Гематологически большая форма β-тал, в дополнение к глубокой анемии, характеризуется типичными изменениями морфологии эритроцитов в виде гипохромии, микроцитоза, анизопойкилоцитоза, полихромазии, фрагментации, базофильной пунктации; в периферической крови присутствует множество нормобластов. Вследствие массивной деструкции эритроидных предшественников в костном мозге, уровень ретикулоцитов обычно невысок. Костный мозг гиперклеточный, с выраженной гиперплазией эритроидного ростка с бедно гемоглобинизированными нормобластами.
При отсутствии заместительной терапии средний уровень Нb составляет 50-60 г/л у пациентов, гомозиготных по β°-тал, и 70 г/л (несколько выше) — у пациентов, гомозиготных по р+-тал. Уровень НЬ А., варьирует в значительных пределах, Hb F всегда повышен и может составлять до 80% общего уровня Нb. Вследствие повышенной продукции и селективной выживаемости F-клеток, Hb F в эритроцитах обычно распределён гетерогенно.
Больные большой формой β-тал нуждаются в заместительной терапии эритроцитарной массой уже с раннего возраста. Адекватный трансфузионный режим обеспечивает им нормальный уровень жизни, блокирует гиперактивность костного мозга, предотвращает развитие деформаций плоских костей и раннего проявления гиперспленизма.
α-Талассемия
α-Талассемия (α-тал) — гемолитическая анемия, вызванная дефицитом синтеза α-глобина в результате потери или повреждения одного или нескольких α-глобиновых генов. Снижение синтеза a-цепей приводит к накоплению свободных γ- и β-цепей и формированию из них тетрамеров-γ4 (Hb Bart's) и нестабильного β4 (Hb Н) с последующим ускорением разрушения эритроцитов. Обладая очень высоким сродством к кислороду, эти тетрамеры не могут выполнять функцию переноса кислорода. Таким образом, клиническая картина тяжёлой α-тал характеризуется комбинацией гипохромной анемии, гемолиза и дефектного транспорта кислорода вследствие различных количеств физиологически неэффективного Нb в эритроцитах. В результате степень тканевой гипоксии значительно превышает ожидаемую при соответствующей степени анемии.
Выделяют 4 группы клинических синдромов α-тал:
• немое носительство;
• α-тал с минимальными изменениями;
• гемоглобинопатию Н;
• α-талассемическую водянку плода.
Тяжесть фенотипического проявления α-тал прямо пропорциональна снижению а-глобинового синтеза.
Немое носительство (α-тал-2 гетерозиготы)
Фенотипически немые носители α-тал мало отличаются от здоровых детей. MCV обычно в пределах 78-80 фл, при этом МСН может соответствовать нижней границе нормы. Все другие гематологические параметры соответствуют норме. Некоторые немые носители могут даже иметь нормальные показатели MCV в пределе 80-85 фл. В крови некоторых из них в период новорождённости выявляют небольшие количества Hb Bart’s (<2%), исчезающего в течение первых месяцев жизни.
Лечение
Трансфузионная терапия
Показания к началу трансфузионной терапии:
• большая форма β-тал, гемоглобинопатия Н при уровне Нb ниже 70 г/л;
• промежуточная и большая формы β-тал, гемоглобинопатия Н при уровне НЬ 70-90 г/л при выраженном отставании физического развития, наличии костных изменений, значительном увеличении селезёнки.
Переливание эритроцитарной массы при талассемии необходимо для поддержания уровня Нb около 120 г/л, что предотвращает развитие вторичного гиперспленизма, деформации костей скелета и гиперволемии за счёт угнетения собственного неэффективного эритропоэза. Интервал между трансфузиями в среднем составляет 2-6 нед и зависит от возможности пациента посещать клинику с определённой частотой, а также от объёма имеющейся эритроцитарной массы.
При отсутствии интеркуррентных заболеваний темп снижения концентрации Нb после переливания составляет примерно 10 г/л в нед. Объём переливаемой эритроцитарной массы определяют в зависимости от интервала между трансфузиями и концентрации имеющейся эритроцитарной массы.
В среднем для повышения уровня Нb пациента на 10 г/л необходимо 3 мл эритроцитарной массы на килограмм массы тела больного при Ht эритроцитарной массы 70%. Железо накапливается в организме медленнее всего при предтранс- фузионном уровне Нb 90-100 г/л и при интервалах между трансфузиями, превышающих 2 нед.
Хелаторная терапия при вторичной (посттрансфузионной) перегрузке железом
Перегрузка организма железом может быть рассчитана по количеству перелитой эритроцитарной массы, насыщению трансферрина железом или уровню СФ. Эти параметры коррелируют как с общими запасами железа в организме, так и с содержанием железа в печени (прямое измерение количества железа в сухом веществе биоптата печени путём спектрометрии атомарной абсорбции). Больные большой формой β-тал в среднем получают 165 (140) мг эритроцитарной массы/кг в год, что соответствует 180 (160) мг Fe/кг в год или 0,49 (0,44) мг Fe/кг в сут (в скобках приведены значения для спленэктомированных больных). Избыток железа накапливается в клетках системы фагоцитирующих макрофагов, их ёмкость составляет около 10-15 г железа; затем железо откладывается во всех паренхиматозных органах и коже, приводя к развитию угрожающих жизни осложнений:
• циррозу печени;
• кардиомиопатии;
• сахарному диабету;
• гипотиреозу;
• гипопаратиреозу;
• гипогонадизму.
Единственная возможность предотвратить поражение органов и тканей — длительное назначение хелатирующих агентов. Для достижения отрицательного баланса железа у трансфузионно-зависимых пациентов необходима экскреция 0,4-0,5 мг/кг железа ежедневно.
Стандартную хелаторную терапию проводят препаратом дефероксамином (Десфералом4 — ДФ) подкожно в дозе 20-40 мг/кг в сут у детей и 40-50 мг/кг в сут у взрослых за 8-12 ч 5-7 дней в нед постоянно или внутривенно непрерывно в течение 24 ч в течение 7 дней с последующим переходом на подкожное введение.
Введение ДФ начинается при уровне ферритина сыворотки (СФ) более 100 нг/мл или железа печени более 3,2 мг/г сухого вещества. Стартовая доза ДФ для детей — 25-30 мг/кг 5 ночей в нед. Терапия осуществляется под контролем терапевтического индекса, который должен поддерживаться не менее 0,025. У детей до 5 лет нежелательно использовать ДФ в дозе более 35 мг/кг, до окончания периода роста - более 50 мг/кг. Если хелаторная терапия начинается до 3-летнего возраста, необходим тщательный мониторинг процессов роста и развития костей.
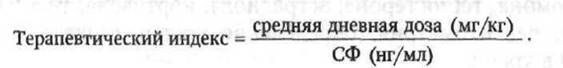
Показания к внутривенному назначению дефероксамина:
• абсолютные;
❖ тяжёлая перегрузка железом;
- СФ постоянно более 2500 мкг/л;
- железо в печени более 15 мг/г сухого вещества;
❖ существенное поражение сердца:
- аритмия;
- сердечная недостаточность;
• дополнительные:
❖ сложности с проведением регулярных подкожных инфузий;
❖ период беременности;
❖ планируемая ТКМ;
❖ активный вирусный гепатит.
Деферазирокс - ещё один пероральный хелатор, который в настоящее время используется в качестве монотерапии первой линии при трансфузионной перегрузке железом у пациентов с талассемией. Рекомендуемая стартовая доза у пациентов с большой формой талассемии — 20 мг/кг однократно в сут с возможным увеличением до 40 мг/кг.
Спленэктомия
Показания к проведению спленэктомии при талассемических синдромах:
• повышение потребности в трансфузиях эритроцитарной массы свыше 200-
220 мл/кг в год (при её Ht 75%);
• спленомегалия, сопровождающаяся болями в левом подреберье и/или угрозой разрыва селезёнки;
• явления гиперспленизма.
Спленэктомию при талассемических синдромах следует проводить по очень строгим показаниям в возрасте не ранее 5 лет после завершения профилактической вакцинации, включающей менингококковую, пневмококковую вакцины, гемофильную вакцину типа В и вакцину против гепатита В (не ранее чем через 2 нед после вакцинации).
Учитывая склонность больных талассемией к гиперкоагуляции, непосредственно перед проведением оперативного вмешательства необходимо исследование гемостаза, назначение профилактических доз ацетилсалициловой кислоты (80 мг/кг в сут) или антикоагулянтов прямого действия в раннем послеоперационном периоде.
Диспансерное наблюдение
Раз в месяц — общий клинический анализ крови.
Раз в квартал — исследование обмена железа, биохимический анализ крови (концентрация глюкозы, мочевой кислоты, мочевины, креатинина, активность щелочной фосфатазы, γ-ГТП, АЛТ, ACT, ЛДГ).
Раз в 6 мес — ЭКГ, ЭхоКГ (измерение объёмов желудочков, сократительной способности миокарда, фракции выброса, систолических и диастолических показателей).
Раз в год — вирусологическое обследование:
• маркёры гепатита В и С, ВИЧ;
• биопсия печени с определением содержания железа в сухом веществе;
• оценка функции эндокринных желёз: определение концентрации свободного Т4, ТТГ, паратгормона, фолликулостимулирующего гормона, лютеинизирующего гормона, тестостерона, эстрадиола, кортизола; тест на толерантность к глюкозе, денситометрия, определение уровня общего и ионизированного кальция в крови.
При выявлении отклонений от нормы следует проводить соответствующее лечение.
Качественные гемоглобинопатии (аномальные гемоглобины)
Клинически значимые аномальные гемоглобины могут быть стабильными (например, Hb S, Hb С, Hb Е) и нестабильными (например, Hb Koln, Hb Volga, Hb Alesha). Наследование аутосомно-доминантное.
СЕРПОВИДНОКЛЕТОЧНАЯ АНЕМИЯ
Серповидноклеточная анемия обусловлена гомозиготностью по Hb S или двойной гетерозиготностью: Нb S-β-талассемия или Hb SC (Hb SE, Hb SD).
Патофизиология серповидноклеточной анемии
Точечная мутация в 6-м кодоне β-глобинового гена (замена валина на глютаминовую кислоту) приводит к изменению свойств белковой молекулы глобина.
• Hb S имеет более отрицательный заряд, чем Нb А, и вследствие этого другую электрофоретическую подвижность.
• Деоксиформа Hb S менее растворима, то есть после передачи атома кислорода Hb S полимеризуется, изменяя форму эритроцитов (в виде серпа); процесс полимеризации Hb S частично обратим.
• Серповидные эритроциты склеиваются, прилипают к поверхности эндотелия сосудов, что нарушает реологические свойства крови, приводя к вазоокклюзивным кризам и инсультам, и быстро разрушаются, приводя к гемолизу.
Гематологические черты заболевания:
• анемия — средней тяжести до тяжёлой, нормохромная, нормоцитарная;
• тест на серповидность положительный;
• ретикулоцитоз;
• нейтрофилёз (достаточно часто);
• количество тромбоцитов часто повышено;
• морфология эритроцитов периферической крови:
❖ серповидные клетки;
❖ высокая полихромазия;
❖ нормобласты;
❖ мишеневидные эритроциты;
❖ тельца Жолли (возможно);
• СОЭ низкая (серповидные эритроциты не могут образовывать «монетные столбики»);
• электрофорез гемоглобина — Hb S движется медленнее, чем Нb А.
Кризы
Вазоокклюзивный (болевой) криз
Самое частое проявление серповидноклеточной анемии. В основном поражаются кости и мышцы. Провоцирующими факторами служат инфекции, дегиратация, холод и гипоксия. Дактилит (синдром рук-ног) — болезненное опухание тыльных поверхностей кистей и стоп — характерен для детей до 5 лет. Костные, клинически сходные с остеомиелитом, чаще начинаются в возрасте 3-4 лет. Абдоминальные симптомы (опоясывающий синдром) развиваются в результате окклюзии сосудов брыжейки и инфаркта печени, селезёнки или лимфатических узлов, в этих случаях необходима дифференциальная диагностика с острым животом. Лёгочный синдром (острый грудной синдром) встречается достаточно часто, в основном у подростков и взрослых, и служит основной причиной хронических заболеваний лёгких и смерти, наступающей в результате прогрессирующей дыхательной недостаточности и множественных инфарктов внутренних органов. Острый грудной синдром необходимо дифференцировать с пневмонией. Лечение симптоматическое (антибактериальная и инфузионная терапия, анальгетики, кислород). Такие факторы, как половая связь, мастурбация, инфекция и локальная травма, способствуют развитию приапизма, в некоторых случаях приводящего к импотенции. Болезненная гематурия умеренной степени развивается вследствие папиллярного некроза почек. Кризы ЦНС могут сопровождаться:
• судорогами;
• менингеальными признаками;
• слепотой;
• ретинопатией;
• головокружением;
• острым нарушением мозгового кровообращения;
• инфарктом мозга.
Частота встречаемости кризов ЦНС составляет 7-29%, средний возраст их развития — 7,7 лет. Высок риск развития субарахноидальных кровоизлияний.
Секвестрационный криз
Наиболее часто локализуется в селезёнке (селезёночная секвестрация), развивается редко, в возрасте 5-24 мес, часто приводит к летальному исходу. Признаки, характерные для клинической картины:
• спленомегалия (сброс большого количества крови в селезёнку);
• внезапная острая боль в животе, сопровождающаяся тошнотой и рвотой;
• резкое снижение уровня НЬ, приводящее к гиповолемическому шоку и смерти.
Печёночная секвестрация проявляется:
• внезапным болезненным увеличением печени;
• выраженным повышением уровня билирубина за счёт его прямой фракции;
• повышением активности трансаминаз (АЛТ, ACT).
Может развиться в любом возрасте при фиброзе селезёнки. Лечение состоит в немедленном восполнении ОЦК и коррекции анемии, а также удалении селезёнки.
Апластический криз
Наиболее часто вызывается парвовирусной инфекций В19. Признаки, характерные для клинической картины:
• резкое глубокое снижение уровня Нb (вплоть до 10 г/л) с отсутствием ретикулоцитов и нормобластов в периферической крови;
• число тромбоцитов и лейкоцитов, как правило, не изменено;
• существенное снижение уровня билирубина в сыворотке крови.
Купируется чаще всего самостоятельно в течение 10 дней. При резком снижении содержания Hb показано переливание эритроцитарной массы.
Гемолитический криз
Сопровождается резкой слабостью, бледностью, иктеричностью склер, могут быть боли в животе. В общем анализе крови отмечается снижение Ht до 15% и ниже, ретикулоцитоз. Через несколько дней гемолиз постепенно прекращается. При тяжёлой анемии показано переливание эритроцитарной массы.
Инсульт
Частое осложнение серповидноклеточной анемии у детей. Развивается вследствие окклюзии крупных сосудов головного мозга, часто нескольких. Вероятность повторных инсультов высока. Регулярные переливания эритроцитарной массы, поддерживающие уровень Hb S не более 30%, существенно снижают риск развития повторных инсультов. При острых нарушениях мозгового кровообращения необходимы срочное обменное переливание с использованием эритроцитарной массы, дегидратация с защелачиванием.
Мегалобластический криз
Вызван повышенной потребностью в фолиевой кислоте в результате усиленного эритропоэза, предотвращается профилактическим приёмом фолиевой кислоты внутрь.
Лечение
Трансфузионная терапия при серповидноклеточной анемии чревата повышением вязкости крови до тех пор, пока уровень Hb S не будет значительно снижен; Ht не должен превышать 25-30% перед началом трансфузии эритроцитарной массы. Неотложное переливание крови показано только при необходимости увеличения транспортной функции крови без выраженного снижения уровня Hb S, например:
• при тяжёлой анемии;
• при секвестрационном кризе;
• при апластическом кризе;
• в случае кровопотери;
• перед оперативным вмешательством.
Постоянные переливания эритроцитарной массы проводит при необходимости снижения уровня Hb S менее 30% (инсульте, иногда при тяжёлом болевом кризе, беременности, перед оперативным вмешательством), для этого переливают эритроцитарную массу из расчёта 10-15 мл/кг каждые 3-4 нед. Обменное переливание быстро нормализует Ht и снижает уровень Hb S. Его проводят по жизненным показаниям:
• при остром грудном синдроме тяжёлого течения;
• при инсульте;
• при артериальной гипоксемии;
• рефрактерном приапизме;
• перед офтальмологической операцией;
• перед ангиографией сосудов головного мозга.
При развитии перегрузки железом проводится хелаторная терапия (см. раздел «Талассемии»),
Для длительного снижения уровня Hb S может быть использована фармакологическая стимуляция синтеза фетального Hb, эффективная примерно в 80% случаев. С этой целью назначают гидроксикарбамид (гидреа*) в дозе 20-30 мг/кг в сутки постоянно, эффект дозозависимый.
Кроме того, применяют трансплантацию костного мозга.
ПРИОБРЕТЁННЫЕ ГЕМОЛИТИЧЕСКИЕ АНЕМИИ
Принято выделять несколько основных групп приобретённых гемолитических анемий:
• аутоиммунные гемолитические анемии, обусловленные продукцией антител против естественных антигенов мембраны эритроцитов;
• токсические (в том числе лекарственные) гемолитические анемии, обусловленные воздействием препаратов, токсинов, ядов и других внешних факторов, способных вызвать гемолиз;
• микроангиопатические гемолитические анемии, обусловленные, как принято считать, повреждением эритроцитов отложениями фибрина в мелких сосудах.
Микроангиопатии встречаются в частности:
• при ДВС-синдроме;
• при ТТП;
• при гемолитико-уремическом синдроме;
• при тяжёлой АГ ;
• при васкулитах;
• при эклампсии;
• при метастазирующих опухолях.
Аутоиммунные гемолитические анемии
Аутоиммунные гемолитические анемии (АИГА) обусловлены выработкой антител против естественных антигенов мембраны эритроцитов с их последующей деструкцией клетками системы мононуклеарных фагоцитов. Реже, если аутоантитела способны активировать комплемент по классическому пути, происходит внутрисосудистый лизис клеток-мишеней. Клиренс «сенсибилизированных» антителами клеток происходит преимущественно в селезёнке, в меньшей степени — в печени и периферических лимфатических узлах.
АИГА классифицируют в зависимости от характеристик опосредующих их аутоантител: температуры, при которой антитела реагируют с эритроцитами, и способности вызывать их агглютинацию и гемолиз. Тепловыми называют антитела, связывающие эритроциты при температуре 36 °С, холодовыми — реагирующие с эритроцитами при температуре не более 26 °С. Антитела, на холоде связывающиеся с эритроцитами, а в тепле — вызывающие гемолиз, называют двухфазными. Если антитела способны только агглютинировать эритроциты, то их называют агглютининами (полными или неполными), если они активируют комплемент и вызывают внутрисосудистый гемолиз, то речь идёт о гемолизинах.
Согласно указанным признакам, выделяют следующие виды АИГА:
• с неполными тепловыми агглютининами;
• пароксизмальную холодовую гемоглобинурию (АИГА с двухфазными гемолизинами Доната-Ландштейнера);
• с полными холодовыми агглютининами.
Изредка тепловые агглютинины могут быть полными и принадлежать к классу IgM. Описаны также случаи комбинированных АИГА с тепловыми и холодовыми антителами, в частности, после инфекционного мононуклеоза, когда вирус Эпстайна-Барр активирует огромный пул В-лимфоцитов, продуцирующих широкий спектр антител.
По этиологии АИГА могут быть идиопатическими или вторичными по отношению к инфекциям, иммунодефицитным синдромам, аутоиммунным заболеваниям, лимфопролиферативным синдромам [хронический лимфоцитарный лейкоз (ХЛЛ), лимфомы], опухолям, экспозиции к медикаментам.
ДИАГНОСТИКА
Диагноз АИГА подтверждают положительной прямой пробой Кумбса (прямой антиглобулиновый тест) либо сенсибилизированной пробой Кумбса (полибреновый тест). Использование в реакции Кумбса антител против IgG, IgM и C3d позволяет определить изотип антител и фиксацию комплемента. При отрицательной пробе Кумбса выявление антител, связанных с эритроцитами, следует принимать во внимание только при наличии других признаков аутоиммунного гемолиза. Непрямая проба Кумбса, выявляющая антиэритроцитарные антитела в сыворотке крови, не имеет отношения к диагнозу АИГА.
АИГА с неполными тепловыми агглютининами — самая частая форма у взрослых и детей, хотя у последних, по некоторым данным, пароксизмальная холодовая гемоглобинурия встречается не менее часто, но реже диагностируется. У детей АИГА с неполными тепловыми агглютининами чаще всего носит характер идиопатической, иммунодефицитные синдромы и СКВ — самые частые причины вторичной АИГА. У взрослых эта форма АИГА часто сопутствует другим аутоиммунным синдромам, ХЛЛ и лимфомам.
Антитела при АИГА с неполными тепловыми агглютининами относят к классу IgG, они не способны связывать комплемент. Соответственно, эритроциты выводятся из кровотока посредством их связывания и эритрофагоцитоза в основном в селезёнке. По специфичности антитела часто направлены против детерминант, ассоциированных с комплексом Rh-антигенов.
Клиническая картина АИГА с неполными тепловыми агглютининами складывается из анемического синдрома (бледности, слабости, сердцебиения) и гипербилирубинемии (желтухи, потемнения мочи, изредка — синдрома сгущения жёлчи: болей в правом подреберье, резкого увеличения печени и жёлчного пузыря, перерастянутых густой слоистой жёлчью). Реже отмечают боли в животе и пояснице, более характерные для внутрисосудистого гемолиза.
К лабораторным характеристикам АИГА относят:
• снижение содержания Нb и Ht;
• гипербилирубинемию;
• повышение содержания ретикулоцитов.
При дебюте гемолиза и при эпизодах его усиления типичен гиперлейкоцитоз нередко до 20-25х109/л с левым сдвигом. При дебюте АИГА иногда регистрируется ретикулоцитопения вследствие быстрого клиренса ретикулоцитов антителами и запаздывания гиперплазии и гиперпролиферации эритроидного ростка костного мозга в ответ на гемолиз. Количество тромбоцитов, как правило, нормальное или незначительно повышено. Снижение концентрации тромбоцитов ниже 100х109/л приводит к необходимости исключать синдром Фишера-Эванса, при котором АИГА сочетается с ИТП. Синдром Фишера-Эванса значительно более устойчив к терапии, чем «простая» АИГА. При дебюте АИГА содержание билирубина повышено как за счёт прямой, так и за счёт непрямой его фракции, в дальнейшем вследствие увеличения экспрессии белка MDR доминирует непрямой билирубин. Длительное повышение концентрации прямого билирубина характерно для массивного гемолиза и развития синдрома сгущения жёлчи. У маленьких детей вследствие резкого относительного преобладания массы функциональной печёночной паренхимы над массой циркулирующих эритроцитов концентрация билирубина может не повышаться даже при выраженном гемолизе.
ЛЕЧЕНИЕ
Агрессивность подхода к лечению АИГА с неполными тепловыми агглютининами зависит от клинической переносимости анемии и темпов падения концентрации Нb. Переносимость анемии в большей степени зависит от выраженности ретикулоцитоза, чем от показателей содержания Нb и Ht, поскольку ретикулоциты очень эффективно отдают кислород периферическим тканям вследствие высокого уровня 2,3-дифосфоглицерата. При выраженном ретикулоцитозе (>10%) дети хорошо переносят даже очень низкие уровни Нb — 35-45 г/л. Если АИГА развилась после инфекционного заболевания, уровень Нb не ниже 55-60 г/л, ретикулоцитоз высок, клиническая переносимость анемии хорошая, и темпы падения гемоглобина не более 10 г/л в нед, то выжидательная тактика может быть оправданна. В таких случаях нередка спонтанная регрессия гемолиза в течение 2-6 мес. В остальных случаях необходимо медикаментозное лечение.
Медикаментозное лечение
Внутривенное введение иммуноглобулинов в дозах 3-5 г/кг (т.е. вдвое-втрое превышающих таковые при ИТП!) достаточно эффективно и применимо у маленьких детей с нетяжело протекающей постинфекционной, или «поствакцинальной», АИГА с неполными тепловыми агглютининами. В остальных случаях основой лечения служат ГК. Стартовая доза преднизолона составляет 2 мг/кг. Такую дозу применяют до нормализации уровня Нb, ретикулоцитоза и билирубина, но не менее месяца. Эффект начального лечения преднизолоном никогда не бывает моментальным: концентрация Нb начинает повышаться через 7-10 дней. В то же время при рецидивах гемолиза, когда гиперплазия эритроидного ростка костного мозга чрезвычайно выражена, подъём уровня НЬ может начаться очень быстро. Нормализация ретикулоцитоза всегда запаздывает по отношению к нормализации концентрации Нb. Если содержание Нb достигает нормальных величин, но ретикулоцитоз остаётся выраженным и проба Кумбса положительна, то говорят о компенсированном гемолизе. Полным ответом считают нормализацию уровня Нb и ретикулоцитов. Полной гематологической ремиссией считают нормализацию уровня Нb и ретикулоцитов при отрицательной пробе Кумбса. После нормализации содержания Нb и ретикулоцитов, сохраняющейся в течение по крайней мере 2 нед, можно приступать к снижению дозы преднизолона. АИГА с неполными тепловыми агглютининами относят к стероидзависимым синдромам, имеющим с тенденцию к рецидивам начиная с определённой дозы препарата. Для преднизолона минимальная пороговая доза обычно составляет 10-20 мг в сут. Соответственно, до 25-30 мг в сут дозу можно снижать достаточно быстро: по 5-10 мг в нед под контролем степени ретикулоцитоза и концентрации эритроцитов. После этого дозу снижают на 1,25-2,50 мг в нед в зависимости от массы тела ребёнка. Проба Кумбса часто остаётся положительной, несмотря на стойкий полный гематологический ответ, что не считают препятствием к снижению дозы и полной отмене преднизолона, однако больные с сохраняющейся положительной пробой Кумбса склонны к рецидивам гемолиза.
Если в течение 2-2,5 мес лечения преднизолоном в дозе 2 мг/кг не достигнуто полной нормализации уровня Нb и ретикулоцитов или если ремиссия заболевания зависит от недопустимо высоких доз преднизолона, необходимо рассмотреть вопрос об альтернативном лечении. Весьма эффективный медикаментозный подход к лечению рефрактерных или стероидзависимых больных — лечение циклофосфамидом. Внутривенное введение 400 мг/м2 циклофосфамида с соответствующей дозой месна раз в 2-3 нед часто приводит к удивительно быстрому купированию гемолиза и нормализации уровня Нb. Обычный курс лечения состоит из 3, максимум 4 введений и не вызывает ранних осложнений в виде нейтропении и геморрагического цистита. В то же время риск позднего канцерогенного действия циклофосфамида делает решение о его применении непростым, особенно для детей. Среди других иммуносупрессантов при АИГА с наибольшим успехом применяют азатиоприн.
Плазмаферез и иммуноадсорбция на колонках со стафилококковым протеином А могут оказать выраженный временный эффект, однако их необходимо сопровождать агрессивной иммуносупрессивной терапией, поскольку эти методы чреваты синдромом рикошета.
Спленэктомию, прежде прочно занимавшую место во второй линии терапии АИГА у детей, по вышеупомянутым причинам сегодня применяют реже. Тем не менее зачастую удаление селезёнки — единственный метод, способный «обуздать» тяжёлый гемолиз. Вопрос об удалении селезёнки решают для каждого больного индивидуально. При выборе решения принимают во внимание:
• возраст больного;
• тяжесть гемолиза;
• доступность, стоимость и побочные явления медикаментозного лечения, необходимого для поддержания парциального или полного ответа.
Пароксизмальную холодовую гемоглобинурию (ПХГ) вызывают антитела класса IgG, связывающиеся с эритроцитами при низкой температуре и активирующие комплемент при температуре тела. Ранее ПХГ чаще всего ассоциировали с поздними стадиями врождённого сифилиса — формой, ныне почти не встречающейся. Сегодня наиболее частая — спорадическая, транзиторная форма ПХГ. У детей ПХГ чаще всего опосредована антителами анти-Р специфичности. Антитела при ПХГ реагируют с эритроцитами при охлаждении и вызывают острейший внутрисосудистый гемолиз с острой гемоглобинурией и поражением почек вплоть до острой почечной недостаточности (ОПН). В клинической картине доминируют боли в животе, лихорадка, бледность с отхождением мочи цвета «вишнёвого сиропа» (по словам мам) и «розового портвейна» (по словам отцов). В моче, постоявшей на воздухе, образуются чёрные хлопья. Нередко развивается тромбоцитопения потребления, поэтому поначалу ПХГ иногда нелегко дифференцировать с гемолитико-уремическим синдромом. ПХГ — самоограниченный синдром, самостоятельно разрешающийся в течение нескольких недель/месяцев. Поскольку аутоантитела класса IgM секретируются В-лимфоцитами, не контролируемыми Т-лимфоцитами, при лечении ПХГ ГК неэффективны. Обычно для лечения ПХГ достаточно не допускать охлаждения ребёнка и проводить грамотную инфузионную терапию во время гемолитического криза. Переливаемую эритроцитарную массу необходимо подогреть до 37 °С.
АИГА с полными холодовыми агглютининами (cold agglutinin disease) у детей встречаются гораздо реже других форм. У взрослых это заболевание обнаруживают нередко: эта форма либо вторична по отношению к лимфопролиферативным синдромам, гепатиту С, инфекционному мононуклеозу, либо идиопатическая. При идиопатической форме анемии, однако, также показано присутствие клональной экспансии популяции морфологически нормальных лимфоцитов, продуцирующих моноклональный IgM. В подавляющем большинстве случаев антитела направлены против углеводных детерминант комплекса I/i поверхности эритроцитов. В 90% случаев антитела специфичны в отношении I, а в 10% — образуются антитела против і. Несмотря на то что при этой форме АИГА антитела реагируют с эритроцитами при низкой температуре и связывают комплемент, явный внутрисосудистый тромбоз является редкостью, а клиренс «сенсибилизированных» эритроцитов опосредуется С3d-рецепторами макрофагов печени и в меньшей степени - селезёнки. Провокацией гемолитического криза часто служит переохлаждение: на прогулках в холодную погоду и на ветру, при купании и т.д. Гемолиз при болезни холодовых агглютининов часто носит подострый характер, без катастрофических падений концентрации Нb. Проба Кумбса при этой форме негативна в реакции с анти-IgG, но положительна в реакции с анти-СЗб. Типична яркая спонтанная агглютинация эритроцитов на стекле. Лечение ГК, циклофосфамидом и интерфероном, а также спленэктомия недостаточно эффективны при АИГА с полными холодовыми агглютининами, и полные ремиссии редки. В связи с этим возникает необходимость поиска и внедрения новых методов медикаментозного, в первую очередь иммуносупрессивного лечения АИГА.
Лечение ритуксимабом (моноклональными антителами к молекуле CD20), уже в течение нескольких лет используемым при лечении онкогематологических и аутоиммунных заболеваний, стало ещё одним эффективным методом консервативного лечения АИГА, хотя вопрос о его месте в настоящее время окончательно не решён. Естественно, пока ритуксимаб не считают препаратом первой линии терапии, однако в последующих линиях его место очевидно. С другой стороны, хорошая эффективность ритуксимаба при болезни холодовых агглютининов, обычно резистентной к стандартной иммуносупрессивной терапии, может вскоре выдвинуть его в первую линию. Показания к назначению ритуксимаба при АИГА:
• аутоиммунные гемолитические анемии, вызванные тепловыми или холодовыми антителами;
• синдром Фишера-Эванса:
❖ при рефрактерности к терапии первой (ГК) и второй (спленэктомия, циклофосфамид, высокие дозы иммуноглобулинов) линии;
❖ при зависимости от высоких (>0,5 мг/кг в сут) доз ГК.
Обычный курс терапии ритуксимабом состоит из 4 введений в разовой дозе 375 мг/м2 с недельным интервалом. Согласно имеющимся данным, на ритуксимаб отвечает 50-80% больных с АИГА. Как правило, параллельно с лечением ритуксимабом рекомендуют применять ГК в прежней дозе, если она составляет не более 1 мг/кг в сут. Другую иммуносупрессивную терапию (например, азатиоприн, циклоспорин) рекомендуют отменить. Однако при катастрофическом гемолизе, непосредственно угрожающем жизни пациента, возможно сочетание ритуксимаба с любыми другими методами терапии (сверхвысокие дозы ГК, циклофосфамид, высокие дозы иммуноглобулина в/в). Как правило, снижение темпа гемолиза и начало повышения уровня Hb наступает со 2-3-й нед терапии, однако качество ответа может значительно варьировать — от полного прекращения гемолиза до его более или менее полной компенсации. Ответившими считают больных, не нуждающихся в гемотрансфузиях и повысивших уровень Нb не менее чем на 15 г/л. Примерно у 25% больных после достижения ремиссии наступает рецидив, как правило, в течение первого года, с большой вероятностью повторного ответа на ритуксимаб. Описаны случаи, когда больные с успехом получали 3 и даже 4 курса ритуксимаба.
Трансфузионная терапия при аутоиммунном гемолизе
Показания к трансфузии эритроцитарной массы зависят не от уровня Нb на данный момент, а от клинической переносимости анемии и от темпов падения содержания Нb. Каждая трансфузия может вызвать внутрисосудистый гемолиз, однако отказ от трансфузии может привести к смерти больного. Необходимо помнить: чем массивнее трансфузия, тем массивнее гемолиз, поэтому цель трансфузии при АИГА — не нормализация концентрации НЬ, а поддержание Ht на клинически достаточном уровне. Минимальное типирование крови для трансфузий при АИГА включает:
• определение АВ0-принадлежности;
• определение полного резус-фенотипа (D, Сс, Ее);
• типирование по антигенам Келла и системы Даффи.
Трансфузии эритроцитарной массы при АИГА связаны с определёнными трудностями. Во-первых, все образцы крови одной группы агглютинируют, соответственно, по классическим канонам, несовместимы. Во-вторых, в Российских клиниках невозможно дифференцировать аллоантитела, развившиеся в результате предыдущих гемотрансфузий и способные вызывать тяжелейший внутрисосудистый гемолиз, от аутоантител, вызывающих внутриклеточный гемолиз. Именно поэтому к трансфузиям рекомендуют относиться максимально консервативно. Для профилактики фебрильных негемолитических реакций рекомендуют лейко-
фильтрацию эритроцитарной массы фильтрами III—IV поколений или в крайнем случае её отмывание. Отмывание эритроцитарной массы не ослабляет гемолиза и не снижает риска образования аллоантител.
Лекарственная гемолитическая анемия
Лекарственная гемолитическая анемия развивается вследствие воздействия многих лекарственных препаратов, вызывающих гемолиз. Известно 3 механизма развития лекарственной (иммунной) гемолитической анемии.
Первый механизм развития гемолиза заключается в том, что препарат вызывает образование антител класса IgG к антигенам эритроцита (часто относящимся к Rh-антигенам). В результате развивается АИГА с тепловыми агглютининами. Подобный механизм образования антиэритроцитарных антител описан при применении многих препаратов, в частности, метилдопы, тенипозида, некоторых НПВП.
Для реализации второго механизма развития гемолиза необходимо связывание лекарственного препарата или его метаболита с мембранными белками эритроцитов, вследствие чего образовавшийся комплекс реагирует с соответствующими антителами. Этот так называемый гаптенный механизм типичен для некоторых антибиотиков (пенициллины, цефалоспорины, тетрациклин), особенно при их применении в высоких дозах.
Третий механизм развития гемолиза связан с тем, что антитела класса IgM реагируют с лекарственным препаратом в кровотоке и образовавшийся иммунный комплекс на короткий срок присоединяется к эритроциту, в результате чего наступает активация комплемента и развивается внутрисосудистый гемолиз.
ЛЕЧЕНИЕ
Лечение лекарственной гемолитической анемии заключается:
• в устранении этиологического фактора (отмене препарата);
• в назначении специфического лечения, направленного на устранение гемолиза;
• в симптоматическом лечении.
Токсические гемолитические анемии
этиология
Гемолиз эритроцитов могут вызывать многие химические вещества и бактериальные токсины. Гемолиз вызывают такие химические вещества, как;
• мышьяковистый водород;
• свинец;
• соли меди (вследствие угнетения активности пируваткиназы и некоторых других ферментов эритроцитов);
• хлораты калия и натрия;
• резорцин;
• нитробензол;
• анилин.
Описаны случаи гемолитической анемии после укуса пчёл, скорпионов, пауков, змей (в частности, гадюк). Очень распространены и опасны отравления грибами, особенно сморчками, чреватые тяжёлым острым гемолизом.
Механизм гемолиза
Механизм гемолиза при токсических гемолитических анемиях может быть различным. Иногда гемолиз развивается вследствие резкого окислительного эффекта (как при энзимопатических анемиях), нарушения синтеза порфиринов, продукции
аутоиммунных факторов и т.д. Чаще всего при токсических анемиях наблюдается внутрисосудистый гемолиз. Гемолитические анемии могут возникать и при инфекционных заболеваниях. Например, малярийный плазмодий способен проникать внутрь эритроцитов, которые затем элиминируются селезёнкой, a Clostridium welchii выделяют α-токсин-лецитиназу, взаимодействующую с мембранными липидами эритроцитов с образованием гемолитически активного лизолецити- на. Возможны и другие варианты: абсорбция бактериальных полисахаридов на эритроцитах с последующим образованием аутоантител, разрушение бактериями поверхностного слоя мембраны эритроцитов и др.
КЛИНИЧЕСКАЯ КАРТИНА
В зависимости от течения выделяют острые и хронические токсические гемолитические анемии. При острых токсических гемолитических анемиях возникает внутрисосудистый гемолиз, проявляющийся гемоглобинемией, гемоглобинурией, а также иногда сопровождающийся явлениями коллапса и анурии. Одна из наиболее ярких моделей острого токсического гемолиза — так называемый гироми- трийный синдром, возникающий вследствие отравления грибами рода Gyromitra из группы сморчковых грибов — строчками (Gyromitra esculenta, Строчок обыкновенный). Помимо острого внутрисосудистого гемолиза (ДВС-синдрома), гироми- трийный синдром включает:
• гастроинтестинальные симптомы, проявляющиеся в первые 6-24 ч после отравления и продолжающиеся от 1 до 3 дней;
• неврологический синдром с астенией и резкой головной болью;
• гипертермию;
• гепатит с выраженным цитолизом.
При этой форме острого гемолиза весьма вероятен летальный исход.
ЛЕЧЕНИЕ
Лечение состоит в прекращении контакта с токсическим агентом или в элиминации такового (в том числе, если возможно, с помощью соответствующего антидота), а при инфекционных заболеваниях — в адекватной антибактериальной или противогрибковой терапии. При тяжёлой анемии показана заместительная терапия. Помимо этого больной нуждается в экстренной посиндромной терапии (лечение почечной недостаточности, гепатита, неврологического синдрома).