Болезни крови у детей и подростков условно возможно разделить на незлокачественные и злокачественные.
Основные группы незлокачественных заболеваний крови у детей и подростков:
• анемии различного генеза;
• болезни лейкоцитов и тромбоцитов;
• геморрагические заболевания и тромбофилии;
• синдромы костно-мозговой недостаточности;
• болезни селезёнки и лимфатических узлов;
• лейкемоидные реакции крови;
• некоторые другие.
Злокачественные болезни крови (ранее называемые гемобласто- зами):
• острые лейкозы;
• хронический миелолейкоз;
• лимфомы;
• гистиоцитозы.
ЭПИДЕМИОЛОГИЯ
Эпидемиология злокачественных болезней крови представлена в главе «Эпидемиология онкологических заболеваний у детей».
В последние годы, по данным официальной статистики Министерства здравоохранения РФ, отмечен рост заболеваемости болезнями крови у детей (рис. 14-1А) и подростков (рис. 14-1Б).
Рис. 14-1. Заболеваемость болезнями крови и кроветворных органов детей в возрасте от 0 до 14 лет (А) и подростков от 15 до 18 лет (Б) в РФ в период с 1992 по 2002 г. (данные официальной статистики Минздрава РФ).
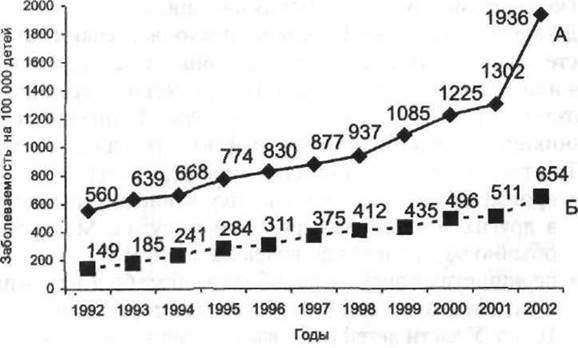
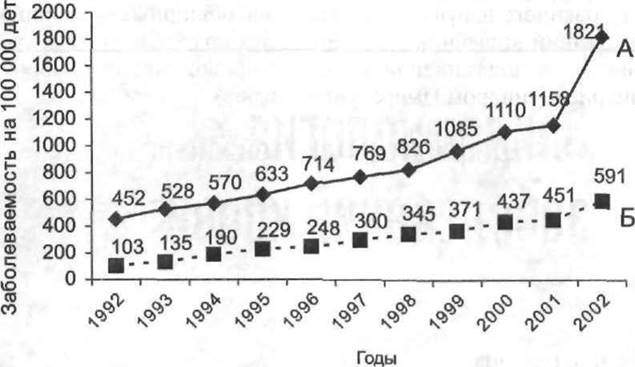
Рис. 14-2. Заболеваемость анемиями детей в возрасте от 0 до 14 лет (А) и подростков от 15 до 18 лет (Б) в РФ в период с 1992 по 2002 г. (данные официальной статистики Минздрава РФ).
АНЕМИИ
Для РФ характерен рост заболеваемости анемией как детей (рис. 14-2А), так и подростков (рис. 14-2Б).
Железодефицитные анемии
КОД ПО МКБ-10
D50. Железодефицитная анемия.
Важная частотная характеристика: 90% анемий у детей — железодефицитные анемии, у взрослых эта цифра достигает 80%. Остальные 10% (у взрослых 20%) приходятся на другие виды анемий: наследственные и приобретённые гемолитические анемии, конституциональные и приобретённые апластические анемии (АА). Истинные цифры заболеваемости и распространённости ЖДА у детей в нашей стране неизвестны, но, скорее всего, достаточно высоки, особенно у детей раннего возраста. Масштабность проблемы можно оценить, рассмотрев данные ВОЗ: 3 600 000 000 человек на земле имеют латентный дефицит железа и ещё 1 800 000 000 человек страдают ЖДА.
Дефицит витамина В12
КОД ПО МКБ-10
D51. Витамин В12-дефицитная анемия.
Дефицит витамина В12 очень редко встречают в педиатрической практике. Системный дефицит витамина В]2 возникает вследствие его нарушенного всасывания или дефицита в пище, например, у детей на грудном вскармливании при недостаточности данного витамина у матери. Тканевый дефицит витамина В12 может возникнуть вследствие редкого дефекта транспортного механизма.
Три типа мальабсорбции витамина В12 у детей:
• врождённое изолированное отсутствие внутреннего фактора при нормальной в других отношениях функции желудка. Межобластная анемия при этом обычно развивается до возраста 2,5 лет;
• пернициозная анемия, подобная этой же болезни у взрослых, с атрофией желудка и антителами против внутреннего фактора в сыворотке крови. Развивается после 10 лет. У части детей развиваются множественные эндокринные нарушения;
• мальабсорбция при нормальной секреции внутреннего фактора вследствие генерализованного нарушения всасывания; обширных операций на кишечнике; заболеваний кишечника; семейного избирательного дефекта всасывания витамина В12 в подвздошной кишке, сопровождаемого доброкачественной протеинурией (синдром Имерслунд-Гросбек).
Дефицит фолиевой кислоты
КОД ПО МКБ-10
D52. Фолиеводефицитная анемия.
Четыре случая развития дефицита фолиевой кислоты в педиатрической практике:
• недоношенные дети;
• инфекции;
• мальабсорбция;
• хронический гемолиз.
Достоверных сведений о заболеваемости дефицитом фолиевой кислоты нет.
Гемоглобинопатии
Генетически детерминированные дефекты белкового синтеза могут быть количественными, влияющими на темпы синтеза цепей глобина (талассемии) или качественными вследствие нарушений структуры цепей (гемоглобинопатии). Наиболее известные гемоглобинопатии (гемоглобин S, С, D, Е) обусловлены замещениями в p-цепях глобина. Чаще всего при рождении ребёнка их не выявляют в связи с тем, что в это время синтез p-цепей незначителен. Замещения в a-цепях можно обнаружить как при рождении ребёнка, так и в дальнейшем, хотя они обычно бессимптомны. Замещения в γ-цепях выявляют только в неонатальном периоде; замещения в 8-цепях не могут быть обнаружены до возраста 5-6 мес, когда в норме впервые появляется гемоглобин А2. Гемоглобинопатии с изменением структуры не β-цепей встречают редко и они представляют в основном теоретический интерес, так как не имеют клинических проявлений (анемии, желтухи и др).
Талассемии
КОД ПО МКБ-10
D56. Талассемия.
Заболевание наиболее распространено в Средиземноморье, Юго-Восточной Азии, Ближнем и Среднем Востоке, государствах Закавказья (Азербайджане, Армении, Грузии), Средней Азии (Таджикистане, Туркмении, Узбекистане), РФ, Дагестане и Поволжье (Татарии, Башкирии). Среди русского населения частота обнаружения гена β-талассемии в гетерозиготном состоянии составляет около 1%; α-талассемию встречают практически в тех же популяциях, что и β-талассемию. Широкая миграция населения привела к увеличению количества больных в нетипичных регионах РФ. Распространению гена талассемии способствуют этнические факторы, обычаи, родственные браки.
Серповидноклеточная анемия
КОД ПО МКБ-10
D57. Серповидноклеточные нарушения.
Серповидноклеточная анемия — аутосомно наследуемое заболевание, вызванное аномальной структурой β-глобина и гомозиготным носительством S-гемоглобина. Серповидноклеточная анемия сочетается с гемоглобинопатиями С, D, Е или талассемией. Заболевание редкое, обычно описывают единичные случаи; встречают у жителей Азербайджана, Грузии, Дагестана.
Ферментопатии
ДЕФИЦИТ ГЛЮКОЗО-6 ФОСФАТДЕГИДРОГЕНАЗЫ КОД ПО МКБ-10
D55.0. Анемия вследствие недостаточности глюкозо-6-фосфатдегидрогеназы [Г-6-ФД].
Дефицит глюкозо-6-фосфатдегидрогеназы (Г-6-ФД) достаточно широко распространён в мире. Примерно 400 000 000 человек являются носителями патологического гена. Описано более 50 вариантов этого фермента среди различных расовых и этнических групп. Среди афроамериканцев частота дефицита Г-6-ФД составляет от 6 до 20-30%; курдских евреев частота носительства патологического гена — 0,7%. В России, по данным популяционного генетического исследования, проведённого в 70-80 гг. XX в., носительство гена составляло среди русских 0,4, татар — 3,3, удмуртов — 2,2, чеченцев — 1,3, народов Дагестана — 5-11,3%. Высокая частота заболевания отмечена среди населения Закавказских государств.
ДЕФИЦИТ ПИРУВАТКИНАЗЫ КОД ПО МКБ-10
D55.2. Анемия вследствие нарушений гликолитических ферментов.
Дефицит пируваткиназы — заболевание, приводящее к развитию хронической несфероцитарной гемолитической анемии. В литературе описано более 400 пациентов с дефицитом пируваткиназы преимущественно из Северной Европы. Описаны случаи болезни среди мексиканцев, итальянцев, японцев, афроамериканцев, сирийцев. Большинство вариантов дефицита пируваткиназы — сложное гетерозиготное носительство, однако описано и гомозиготное носительство, вызванное чаще всего близкородственными браками. К настоящему времени выявлено 62 генные мутации, затрагивающие в основном места присоединения фермента к субстрату.
ДЕФИЦИТ ГЛЮКОЗОФОСФАТИЗОМЕРАЗЫ КОД ПО МКБ-10
D55.1. Анемия вследствие других нарушений глутатионового обмена.
Дефицит глюкозофосфатизомеразы занимает третье место среди энзимопатий после дефицита Г-6-ФД и пируваткиназы. В литературе описано более 40 семей с данным заболеванием. Частота гетерозиготного носительства в большинстве популяций не изучена, существуют данные только японских исследователей, показавших распространённость в 2,3 случаях на 1000 населения.
Мембранопатии
НАСЛЕДСТВЕННАЯ СФЕРОЦИТАРНАЯ ГЕМОЛИТИЧЕСКАЯ АНЕМИЯ (БОЛЕЗНЬ МИНК0ВСКОГО-ШОФФАРА)
КОД ПО МКБ-10
D58. Другие наследственные гемолитические анемии.
Наследственная сфероцитарная гемолитическая анемия — заболевание, относящееся к группе наследственных анемий, в основе которых лежит молекулярный дефект белков мембраны эритроцита.
Виды дефектов белков мембраны эритроцитов:
• изолированный дефект спектрина;
• комбинированный дефект спектрина и анкирина (30-60% случаев);
• парциальный дефицит белка полосы 3 (15-40% случаев);
• дефицит белка 4.2;
• дефицит других мембранных белков.
Распространённость наследственной сфероцитарной гемолитической анемии среди жителей Северной Европы — 1 случай на 5000 человек. Примерно с такой же частотой наследственную сфероцитарную гемолитическую анемию встречают в США — 22 случая на 100 000 населения. Заболевание одинаково часто развивается у лиц мужского и женского пола. Наследуется по доминантному типу, однако степень экспрессии гена вариабельна, в связи с чем создаётся впечатление, что болезнь «перепрыгнула» через поколение; кроме того, в одной семье тяжесть течения процесса может быть различной.
НАСЛЕДСТВЕННЫЙ ЭЛЛИПТОЦИТОЗ (ОВАЛОЦИТО3)
КОД ПО МКБ-10
D58.1 Наследственный эллиптоцитоз.
Наследственный эллиптоцитоз — заболевание, в основе которого также лежит дефект мембраны эритроцитов.
Причины развития наследственного эллиптоцитоза:
• мутации α- и β-спектри на;
• мутация белка 4.1;
• мутация гликофорина С.
По-видимому, болезнь повсеместна. Распространённость хорошо изучена в США и составляет 3-5 случаев на 10 000 населения. В районах, эндемичных по малярии, она значительно выше: в Африке, например, достигает 0,6% всего населения.
Анемия при хронических болезнях
КОД ПО МКБ-10
D63.8. Анемия при других хронических болезнях, классифицированных в других рубриках.
Анемия при хронических болезнях — заболевание, сопровождающее болезни соединительной ткани, сепсис, остеомиелит, туберкулёз и др. Анемию при хронических болезнях почти не учитывают как самостоятельное заболевание; сведений о её распространённости нет. При ряде хронических заболеваний частота встречаемости достигает 100%.
АНЕМИЯ ПРИ ХРОНИЧЕСКОЙ ПОЧЕЧНОЙ НЕДОСТАТОЧНОСТИ И В ПРОЦЕССЕ ДИАЛИЗНОГО ЛЕЧЕНИЯ
КОД ПО МКБ-10
D63. Анемия при хронических болезнях, классифицированных в других рубриках.
Преддиализная стадия ХПН сопровождается анемией в 60-80% случаев, в терминальной стадии ХПН анемию выявляют в 100% случаев.
Тромботическая тромбоцитопеническая пурпура
(синдром Мошкович)
КОД ПО МКБ-10
М31.1. Тромботическая микроангиопатия.
Тромботическая тромбоцитопеническая пурпура (ТТП) — редкое заболевание. Основные патофизиологические механизмы:
• множественное микротромбообразование;
• интенсивная агрегация тромбоцитов;
• диссеминированная закупорка мелких артерий и артериол пластиночными тромбами;
• вторичный неиммунный гемолиз;
• тромбоцитопения потребления;
• ишемическое поражение мозга, почек, печени, сердца и других органов.
В отличие от гемолитико-уремического синдрома при ТТП нет связи с инфекцией; обычно болезнь развивается у детей старше 3 лет. Сведений о заболеваемости нет.
Семейный наследственный эритроцитоз
КОД ПО МКБ-10
D75.0. Семейный эритроцитоз.
Семейный наследственный эритроцитоз — заболевание, характеризующееся увеличением массы эритроцитов и повышением уровня гемоглобина. В России эндемический очаг семейного наследственного эритроцитоза выявлен Л.А. Поляковой в 1997 г. на территории Чувашии, причём только в популяции чувашей. Распространённость семейного наследственного эритроцитоза среди детей и подростков от 0 до 18 лет на территории Чувашской Республики составляет 11,8 на 100 000 детей. Заболевание регистрируют среди девочек в 1,5 раза чаще.
Апластические анемии
КОД ПО МКБ-10
D61. Другие апластические анемии.
Виды АА:
• врождённые [анемия Фанкони (АФ), анемия Даймонда-Блекфена (АДБ), врождённый дискератоз, анемия Швахмана-Даймонда-Оски, амегакариоцитарная тромбоцитопения];
• приобретённые (идиопатические, вызванные вирусами, лекарствами или химическими веществами).
АА встречают с частотой 1-2 случая на 1 000 000 населения в год и считают редкими болезнями крови. Приобретённые АА развиваются с частотой 0,2-0,6 случая на 100 000 детей в год. Среднегодовой показатель заболеваемости АА у детей в период с 1979 по 1992 г. в Республике Беларусь - 0,43±0,04 на 100 000 детей. Разницы в показателе заболеваемости АА у детей до катастрофы на Чернобыльской АЭС и после не получено.
АНЕМИЯ ДАЙМОНДА-БЛЕКФЕНА КОД ПО МКБ-10
D61.0. Конституциональная апластическая анемия.
АДБ описана под многими названиями: парциальная красноклеточная аплазия, врождённая гипопластическая анемия, истинная эритроцитарная анемия, первичная красноклеточная болезнь, эритрогенезис имперфекта. Заболевание редкое, L.K. Diamond и соавт. в 60-е гг. XX в. описали всего 30 случаев этой болезни, к настоящему времени описано более 400 случаев.
Длительное время считали, что заболеваемость АДБ составляет 1 случай на 1 000 000 живых новорождённых. В 1992 г. L. Wranne сообщил о более высокой заболеваемости — 10 случаев на 1 000 000 новорождённых. Показатель заболеваемости АДБ по данным французского и английского регистров составляет 5-7 случаев на 1 000 000 живых новорождённых. Соотношение по полу почти одинаково. Более 75% случаев АДБ — спорадические; 25% семейный характер, причём в некоторых семьях зарегистрировано несколько больных. Регистр больных АДБ США и Канады включает 264 больных в возрасте от 10 мес до 44 лет.
АНЕМИЯ ФАНКОНИ КОД ПО МКБ-10
D61.0. Конституциональная апластическая анемия.
АФ — редкое аутосомно-рецессивное заболевание, характеризующееся множественными врождёнными физическими аномалиями, прогрессирующей костномозговой недостаточностью и предрасположенностью к развитию злокачественных новообразований. Заболеваемость АФ — 1 случай на 360 000-3 000 000 населения. Заболевание распространено среди всех национальностей и этнических групп. Минимальный возраст проявления клинических признаков — период новорождённости, максимальный — 48 лет. В регистре больных АФ НИИ детской гематологии М3 РФ зафиксированы данные 69 больных. Средний возраст манифестации заболевания — 7 лет (2,5-12,5 лет). Выявлено 5 семейных случаев.
ГЕМОРРАГИЧЕСКИЕ ЗАБОЛЕВАНИЯ Пурпура и другие геморрагические состояния
ИДИОПАТИЧЕСКАЯ ТРОМБОЦИТОПЕНИЧЕСКАЯ ПУРПУРА КОД ПО МКБ-10
D69.3. Идиопатическая тромбоцитопеническая пурпура.
Идиопатическая тромбоцитопеническая пурпура (ИТП), по мнению многих гематологов,— часто встречаемое геморрагическое заболевание. Однако в единственном в нашей стране исследовании показано, что показатель заболеваемости ИТП составляет в Челябинской области 3,82±1,38 случая на 100 000 детей в год и не имеет тенденции роста.
ТРОМБОЦИТОПАТИИ КОД ПО МКБ-10
D69.1. Качественные дефекты тромбоцитов.
Тромбоцитопатии — группа заболеваний, при которых геморрагический синдром обусловлен качественной неполноценностью тромбоцитов при нормальном или несколько сниженном их количестве. Различают наследственные и приобретённые (вторичные) тромбоцитопатии. Полагают, что тромбоцитопатии имеют большую распространённость: болеет каждый десятый человек в мире. Дети с наследственными тромбоцитопатиями составляют 60-80% общего количества больных с наследственными нарушениями системы гемостаза. В работах последних лет показано, что классические наследственные тромбоцитопатии у детей в 24% случаев связаны с наследуемыми дефектами рецепторов, ферментов и морфологии тромбоцитов, а в 76% случаев определяются наследственными дефектами строения коллагена, нарушениями фосфорно-кальциевого и других видов обмена.
Гемофилия
КОД ПО МКБ-10
D66. Наследственные дефицит фактора VIII.
D67. Наследственный дефицит фактора IX.
Заболевание встречают с частотой 1-2 случая на 10 000 новорождённых мальчиков в год. Соотношение гемофилии А (дефицит фактора VIII) и гемофилии В (дефицит фактора IX) — 4:1. Заболеваемость гемофилией мальчиков в РФ имеет тенденцию к росту за счёт появления так называемых спорадических случаев гемофилии. Эти случаи объясняют спонтанными генетическими мутациями или реализацией скрытого гетерозиготного носительства патологического гена.
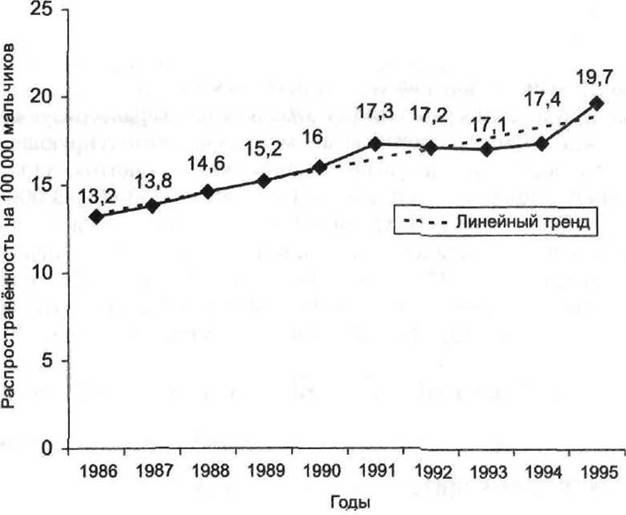
Рис. 14-3. Динамика показателя распространённости гемофилии типа А и В у мальчиков Москвы (суммарно) за период с 1986 по 1995 г. и его линейный тренд (на 100 000 мальчиков в возрасте от 0 до 14 лет).
Повышение качества специализированной медицинской помощи больным гемофилией привело к росту показателя распространённости гемофилии и крайне низкой смертности от этой болезни в детском возрасте. По данным Детского гематологического центра при Измайловской детской клинической больнице, распространённость гемофилии в Москве имеет явную тенденцию к росту (рис. 14-3).
Болезнь Виллебранда
КОД ПО МКБ-10
D68.0. Болезнь Виллебранда.
Болезнь Виллебранда (БВ) — самое частое наследственное геморрагическое заболевание в мире. По данным эпидемиологического исследования, объединившего наблюдения за больными БВ в нескольких сотнях гематологических отделений и центров США, Европы и Азии, заболеваемость тяжёлыми формами болезни составила 1,38-1,53 случая на 1000 000 населения. Е. Werner и соавт. (1993) обследовали 600 здоровых детей в 3 американских штатах.
Критерии постановки диагноза (Werner Е. и др.):
• наличие симптомов кровоточивости у больного:
• снижение фактора Виллебранда у больного;
• наличие симптомов кровоточивости хотя бы у одного близкого родственника.
Диагноз БВ установлен у 8 (1,3%) детей. К. Shinmyozu и соавт. (1991) в своём
исследовании показали, что распространённость бессимптомного снижения уровня фактора Виллебранда в популяции составляет 1,3%. Таким образом, популяционные исследования показали, что приблизительно 1-1,5% населения земного шара могут страдать БВ. Нескольких типов БВ (типы I, ІІА, ІІВ, ІІМ, ІІN, III), субклиническое течение процесса у значительной части больных, трудности лабораторной диагностики (необходимость определения фактора Виллебранда и его мультимеров и антигенов, фактора VIII и др.) приводят к тому, что заболевание в ряде случаев не диагностируют. Первичное выявление заболевания при травмах и проведении операций представляет большую опасность для больного.
В Москве среднегодовой показатель заболеваемости БВ составил в период с 1978 по 1995 г. 0,78±0,1 случая на 10 000 живых новорождённых мальчиков и девочек. Среднегодовой показатель распространённости БВ в г. Москве за эти же годы - 10,06±1,79 случая на 100 000 детского населения. В детской практике почти отсутствует смертность от БВ; за 18 лет наблюдения зарегистрирован один случай смерти больного, в связи с чем показатель смертности составил 0,0078 на 10 000 мальчиков и девочек, рождённых живыми.
Нейтропении
КОД ПО МКБ-10
D70. Агранулоцитоз.
Нейтропении - заболевания, выявляемые преимущественно у детей до 3 лет. Заболеваемость аутоиммунной нейтропенией, в дальнейшем имеющей хроническое течение, составляет более 1 случая на 1 000 000 детей в год.
Лимфоаденопатии и регионарные лимфадениты
КОД ПО МКБ-10
D75. Другие болезни крови и кроветворных органов.
Принято считать, что лимфоаденопатии и лимфадениты встречают в педиатрической практике часто. Необходимо иметь в виду, что в кабинете врача-педиатра, а тем более врача-гематолога (онколога) происходит концентрация больных, в том числе и с увеличенными лимфатическими узлами, то есть приходится иметь дело с так называемой госпитальной выборкой. Обследование 1607 детей в возрасте
6-16 лет, осмотренных преимущественно в весенне-летний период вне эпидемии респираторных заболеваний, показало, что частота встречаемости лимфоаденопатий - 1,9%, лимфаденитов — 1,5%. По данным НИИ детской онкологии и гематологии РОНЦ РАМН, из 678 больных Москвы, направленных для обследования по поводу подозрения на злокачественное новообразование лимфоидной и кроветворной системы в течение одного года, опухолевые заболевания выявлены у 71 больного (10,6%). У остальных больных выявлены реактивные лимфоаденопатии, связанные с острым и хроническим воспалением или другими причинами. Таким образом, количество больных с увеличенными вследствие пролиферативного заболевания лимфатическими узлами составляет в общей популяции детей всего 1% и увеличивается до 10% благодаря искусственной концентрации в специализированном онкогематологическом учреждении.
Лейкемоидные реакции крови в зависимости от гиперпродукции клеток в периферической крови разделяют на нейтрофильные, эозинофильные, лимфоцитарные и моноцитарные. Наиболее часто развивается лейкемоидная реакция крови нейтрофильного типа. Любой так называемый сдвиг формулы крови влево при воспалении — не что иное, как лейкемоидная реакция крови нейтрофильного типа. Распространённость этой лейкемоидной реакции крови неизвестна, скорее всего, велика. На втором месте по частоте встречаемости находится лейкемоидная реакция крови эозинофильного типа.
Причины лейкемоидных реакций крови эозинофильного типа:
• паразитарные заболевания;
• аллергические реакции.
В результате анализа гемограмм 1321 ребёнка в возрасте от 6 мес до 14 лет из г. Клинцы и Клинцовского района Брянской области выявлено, что повышенное количество эозинофилов (более 6%) в периферической крови зарегистрировано у 26,3% детей; у 20,6% обнаружили лейкоцитоз (более 10,0х109/л), что позволило диагностировать у них лейкемоидную реакцию крови эозинофильного типа.
Причины эозинофилии:
• паразитозы (33,4%);
• аллергические состояния (5,2%);
• хронические очаги инфекции (0,98%).
У детей, проживающих в городе, эозинофильные реакции крови встречают в 2 раза чаще, чем у сельских детей, что объясняется неблагоприятным экологическим воздействием.