ГКЛ — группа конституционально обусловленных заболеваний неясной этиологии с выраженной пролиферацией клеток моноцитарно-макро- фагальной системы и клеток дендритной системы в различных органах и тканях.
Частота ГКЛ составляет примерно 1 случай в год на 100 тыс. детского населения в возрасте до 1 года и 2 больных в год на 1 млн детей в возрасте до 15 лет [Миллер Д. Р., 1990].
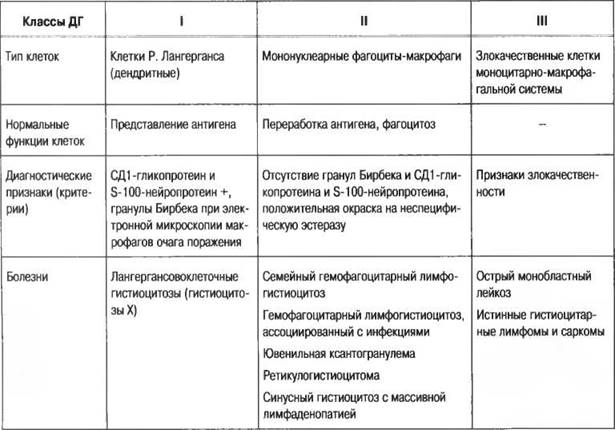
Согласно рекомендациям Международной группы экспертов по ГКЛ (1987) следует различать следующие формы (табл. 207).
Гистиоцитозы X — группа заболеваний (в настоящее время по инициативе С. Nezelof (1973) их называют Лангергансовоклеточный гистиоцитоз — ЛКГ или гистиоцитоз из клеток Лангерганса), общим патогенетическим звеном которых является клональная реактивная пролиферация клеток с фенотипом эпидермальных клеток Лангерганса (отростчатых клеток), имеющих гранулы Бирбека при электронной микроскопии или CD 1-гликопротеин и S-100-нейропротеин при исследовании с помощью моноклональных антител.
Таблица 207
Классификация гистиоцитозов
Вторично в вакуолях цитоплазмы клеток наблюдается накопление продуктов нарушенного обмена липидов, холестерина, придающих узелкам пролиферации желтый цвет, что и дало название для одного из вариантов болезни — ксантоматоз. Клетки Лангерганса происходят из костного мозга и длительность их пребывания в коже составляет около 3 нед, являются антиген-презентующими клетками эпидермиса, продуцируют интерлейкин-1, индуцируя Т-клеточную активацию, отторжение трансплантата; опосредуют иммунный ответ при атопическом дерматите, несут на своей поверхности СЗ- и Fc IgG-рецепторы, HLA-DR и CD1.
В XX веке различали три формы течения Л КГ: болезнь Абта—Леттерера—Сиве, болезнь Хенда—Шюллера— Крисчена (ксантоматоз), эозинофильная гранулема (болезнь Таратынова), которые отличаются по клинической картине и прогнозу, но описаны взаимные переходы эозинофильной гранулемы в болезнь Хенда—Шюлл ера—Крисчена, а затем в болезнь Абта—Леттерера—Сиве. Согласно предложению Международного общества гистиоцитологов, выделяют следующие формы ЛКГ:
Поражение одной системы:
— одиночные очаги: одиночные костные очаги, изолированное поражение кожи, одиночный лимфатический узел;
— множественные очаги: множественные костные очаги, множественное поражение лимфатических узлов.
Мультисистемный процесс, множественное поражение органов с нарушением их функций или без него. «Органами риска» являются: печень, легкие, кроветворная система, селезенка.
Этиология и патогенез Л КГ пока не ясны. Болезнь не контагиозна; после введения материала из очагов поражения животным никаких патологических изменений у них не обнаружено. Маловероятно, что это опухоль, так как возможна спонтанная резорбция очагов поражения; более того, она типична при поражении лишь костей и длительном течении. Наиболее убедительным представляется мнение, что Л КГ — иммунопатологический процесс с последующим цитокинопосредованным ЛКГ. Действительно, у ряда больных отмечен дефицит Т-супрессоров (СБ8-лимфоциты), кортикальная атрофия в лимфатических узлах, дисплазия тимуса. У таких больных, наблюдавшихся Д. Р. Миллером (10 из 17 с ксантоматозом), достигнута полная ремиссия при лечении гормонами тимуса. При болезни Абта—Леттерера—Сиве у детей имеется глубокий комбинированный иммунодефицит с более значительным дефектом клеточного звена. В этих случаях помочь может лишь трансплантация костного мозга и иммуносупрессивная терапия.
Гемофагоцитарный лимфогистиоцитоз разделяют на первичный (семейный и спорадический) и вторичный или гемофагоцитарные синдромы, ассоциированные с инфекциями (преимущественно вирусными), опухолями, иммунодефицитами, иммуноопосредованными заболеваниями. Семейный гемофагоцитарный лимфогистиоцитоз — аутосомно-рецессивно наследуемое заболевание, связанное с локусами на хромосомах 9 (9q21.3-22, FHL1) и 10 (10q21-22,FHL2), клинически проявляющееся обычно в течение первого года жизни и без лечения заканчивающееся летально.
Этиология и патогенез III класса ДГ — аналогичны лейкозам.
Болезнь Абта—Леттерера—Сиве развивается в большинстве случаев в раннем возрасте, преимущественно на первом году жизни. Заболевание может начинаться исподволь, с вялости, анорексии, снижения прибавки массы тела, поражения кожи (себорейный дерматит) и слизистых оболочек, субфебрилитета, а также остро, с септической лихорадки. Для периода выраженных явлений болезни характерны:
1) периодическая лихорадка септического типа;
2) изменения на коже (помимо себорейного дерматита, наблюдаются розовые папулезные высыпания в области грудины, позвоночника, покрывающиеся желтоватыми корочками, пятнисто-мелкоточечные геморрагии);
3) гепатоспленомегалия с генерализованным увеличением периферических лимфатических узлов;
4) интерстициальные поражения легких (милиарные очаги, образующие нежную сеть, распространяющуюся на оба легочных поля), иногда очаги деструкции;
5) неправильной формы деструктивные очаги в плоских костях, напоминающие при рентгенологическом исследовании географическую карту, а клинически проявляющиеся как припухлость черепа разной консистенции;
6) отиты, иногда мастоидиты, несахарное мочеизнурение, экзофтальм.
При анализе периферической крови отмечают тромбоцитопению, анемию, повышенную СОЭ, лейкоцитоз с увеличением количества нейтрофилов, плазматических и ретикулярных клеток. Очень часто наслаивается вторичная инфекция и может развиться сепсис.
Болезнь Хенда—Шюллера—Крисчена может возникнуть у детей любого возраста старше года. В типичных случаях наблюдают триаду симптомов: дефекты костей черепа, экзофтальм, несахарный диабет. Однако эта характерная триада симптомов развивается не в первые месяцы болезни и не у всех больных. Поражение костей черепа (клинически — мягкоэластическая припухлость, рентгенологически — причудливой формы очаги деструкции, напоминающие географическую карту) — наиболее постоянный симптом. Наряду с этим у больного наблюдают в различных комбинациях следующие поражения:
1) аналогичные происходящим в черепе изменения в других плоских костях, позвоночнике;
2) экзофтальм (рис. 103) и периорбитальные поражения кожи;
3) несахарное мочеизнурение;
4) задержка роста, физического и полового развития;
5) гепатомегалия, лимфаденопатия, реже ге- патоспленомегалия;
6) желтоватые ксантомные очаги на коже, сочетающиеся с геморрагиями;
7) папулезные и себорейные высыпания на коже головы, спины;
8) стоматиты, рецидивирующие отиты;
9) легочные инфильтраты.
В периферической крови при морфологическом исследовании отмечают лейкоцитоз, повышенную СОЭ, иногда эозинофилию, тромбоцитопению.
Эозинофильная гранулема (болезнь Таратынова) диагностируется чаще у детей дошкольного и школьного возраста и характеризуется слабостью, повышенной утомляемостью, снижением аппетита, болями в костях (нередко после травмы), гнейсом, себорейным дерматитом. Поражаются плоские (череп, таз, ребра) и трубчатые (бедро, голень, плечо) кости, а также позвоночник. На рентгенограмме очаг деструкции имеет округлую, овальную, реже неправильную форму с полициклическим фестончатым контуром без окружающего склероза.
Иногда заболевание протекает бессимптомно, и очаг деструкции обнаруживают случайно, при рентгенологическом исследовании. При анализе периферической крови у больных находят увеличенную СОЭ, реже эозинофилию. Несмотря на то, что эозинофильная гранулема может самопроизвольно исчезнуть без лечения, у некоторых больных могут появиться несахарное мочеизнурение, экзофтальм, очаги деструкции в других костях, что сопровождается гепатомегалией, анемией, изменениями на коже. Наш опыт свидетельствует о возможности развития аутоиммунной гемолитической анемии у больных с эозинофильной гранулемой, причем это может быть одним из первых признаков заболевания.
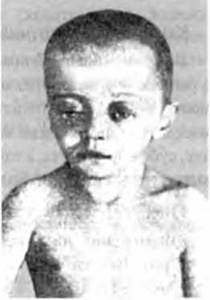
Рис. 103. Экзофтальм у больного с ксантоматозом.
Гемофагоцитарный лимфогистиоцитоз характеризуется:
1) лихорадкой в течение 7 дней и более;
2) сплено- и/или гепатомегалией с повышением уровня сывороточных трансаминаз и билирубина;
3) гипертриглицеринемией;
4) панцитопенией (нормоцитарная анемия с адекватным ретикулоцитозом, тромбоцитопения, нейтропения, но лимфоцитоз с повышением числа активированных Т-клеток);
5) гиперцитокинемией![]()
6) гипофибриногенемией и нередким развитием ДВС-синдрома;
7) транзиторной эритематозно-папулезной сыпью в момент лихорадки;
8) симптомами поражения ЦНС (признаки повышения внутричерепного давления, менингеальные симптомы, атаксия геми- и тетраплегии и др.);
9) повышением количества зрелых гистиоцитов в костном мозге свыше 3% при отсутствии признаков гипоплазии костномозгового кроветворения;
10)выраженным гемофагоцитозом гистиоцитов или в костном мозге, или в селезенке, или в лимфатических узлах.
Диагноз и дифференциальный диагноз
Диагноз прежде всего основывают на характерных клинических, рентгенологических и гематологических данных. Для подтверждения диагноза гистиоцитозов целесообразно произвести миелограмму, биопсию кожи, лимфатического узла, пункцию пораженного участка кости черепа, биопсию очага деструкции кости. Для подтверждения диагноза выявляют гранулы Бирбека при электронной микроскопии, CD-гликопротеин в очагах поражения. Гистиоцитозы в зависимости от клинической картины заболевания дифференцируют от остеомиелита, костного туберкулеза, нейробластомы, первичных и метастатических опухолей мозга, остеокластической саркомы, фиброзной остеодистрофии, лимфогранулематоза, лейкоза, болезней Гоше, Ниманна—Пика, портальной гипертензии с внепеченочным блоком.
Зависит от течения болезни. При остро текущих формах с генерализованным поражением внутренних органов применяют винбластин с преднизолоном. Винбластин вводят внутривенно 1 раз в неделю в дозе 6 мг/м2, преднизолон внутрь ежедневно по 40 мг/м2. Длительность терапии определяется ее эффективностью, но обычный курс индукции ремиссии — 7 нед, и далее переходят на поддерживающую терапию в течение 1 года: 6-мер- каптопурин 50 мг/м2 + преднизолон (40 мг/м2) в 1-5-й дни недели и винбластин (6 мг/м2) в 1-й день недели. При раннем выявлении резистентности к терапии вливания винбластина сочетают с введением вепезида (этопози- да) 150 мг/м2.
Предлагают схемы комбинированной терапии с применением дексаме- тазона, антилимфоцитарного глобулина, вепезида и циклоспорина. Некоторые авторы используют в лечении моноклональные антитела к CD 1-антигену, циклоспорин А ИФа, аналоги нуклеозидов, антитимоцитарный глобулин, талидомид.
При выявлении дефицита СБ8-лимфоцитов показано лечение гормонами тимуса.
При эозинофильной гранулеме и болезни Хенда—Шюлл ера—Крисчена, протекающей лишь с поражением костей и/или несахарным диабетом, необходимости в таком массивном лечении, как правило, нет. В то же время, изолированное применение глюкокортикоидов при любых формах гистиоцитоза X считают неадекватной терапией.
При очаговых поражениях костей и на область турецкого седла при несахарном диабете применяют лучевую терапию. Несахарное мочеизнурение — показание к лечению адиурекрином или десмопрессином.
При ограниченных кожных поражениях применяют мази с глюкокортикоидами, дексаметазоном, мустаргеном или PUVA-терапия — ультрафиолетовое облучение (320-340 нм) в сочетании с фотосенсибилизатором псораленом, даваемым в дозе 0,6 мг/кг за 1-2 ч до УФ (сеансы 3 раза в неделю).
При первичном гемофагоцитарном лимфогистиоцитозе максимальный эффект достигается при трансплантации аллогенного костного мозга или стволовых клеток. Разные авторы указывают на положительный эффект и достижение ремиссии у большинства больных при назначении 5-дневного курса антитимоцитарного глобулина с последующим — циклоспорина А, комбинированной терапии винбластином или этопозидом в сочетании с дексаметазоном, курсов вепезида, винбластина и преднизолона. Лечение вторичных гемофагоцитарных лимфогистиоцитозов предусматривает применение этиотропной терапии.
В частности, при доказанных вирусных инфекциях используют специфические антивирусные препараты, иммуноглобулин внутривенно, для иммуномодуляции — рекомбинантные ИЛ-6, ИФа, И Фу иногда в комбинации с циклоспорином А, колониестимулирующими факторами (КСФ-Г).
Определяется степенью генерализации процесса. Считают, что при отсутствии поражения внутренних органов даже при наличии генерализованного поражения костей прогноз благоприятный. При эозинофильной гранулеме прогноз, как правило, тоже благоприятный, а при болезни Абта—Леттерера—Сиве все же чаще неблагоприятный. Общий процент выздоровлений при всех формах гистиоцитоза X — 70%.