7.1 Введение
Половая дифференцировка плода, которая определяет, будут ли половые органы у ребенка развиты по мужскому или женскому типу, происходит в течение первых недель эмбрионального периода. Позднее, уже в период полового созревания, под влиянием половых гормонов формируются вторичные половые признаки, и девочка превращается в женщину, способную к деторождению. Диагностика нарушений этого процесса требует знания физиологии процесса половой дифференцировки и полового развития.
7.2. Физиологические основы
Половые гормоны
Биосинтез половых гормонов
Первой ступенью биосинтеза половых гормонов является транспорт холестерина с наружной поверхности митохондриальной мембраны на внутреннюю с помощью стероидогенного острого регуляторного белка, или СОРБ (Bose et al., 1996). Здесь холестерин с помощью фермента CYP11A1 (цитохром P450scc) превращается в прегненолон (рис. 7-1) (Hall, 1985). Активность СОРБ регулируется гормонами гипофиза: ЛГ, ФСГ, АКТГ (Stocco u. Clark, 1997). Синтез стероидных гормонов регулируется количеством имеющегося для этого субстрата.
Путем дальнейшего каскада реакций прегненолон превращается в андрогенные или эстрогенные гормоны. Прогестагены сначала под действием фермента CYP17 (17в-гидроксилаза, 17,20- лиаза) путем ступенчатого гидроксилирования превращаются в андрогенный гормон андростендион. Андростендион затем превращается с помощью фермента 17р-гидроксистероиддегид- рогеназы (17р-ГСД) в сильнодействующий андрогенный гормон тестостерон или под действием ароматазы — в эстрогенный гормон эстрон. Сильнодействующий эстрогенный гормон 17(3- эстрадиол образуется из эстрона под действием 17(3-ГСД или из тестостерона под действием ароматазы. Эта ароматаза (CYP19) локализуется в клетках фолликулов яичников и активируется под влиянием ЛГ (см. рис. 7-1).
Половая дифференцировка
Генетический пол
Генетический пол определяется половыми хромосомами.
При оплодотворении яйцеклетки сперматозоидом, который содержит гаплоидный набор с X- или Y-хромосомой, эмбрион получает женский (46,XX) или мужской (46,XY) набор хромосом. На Y-хромосоме локализуется ген. характеризующийся высокой консервативностью [так называемая область Y-хромосомы, детерминирующая пол (SRY — Sex-determinierende Region des Y- Chromosoms), или фактор, детерминирующий развитие яичек (TDF — Testis-determinierende Faktor) (Sinclair et al. 1990).
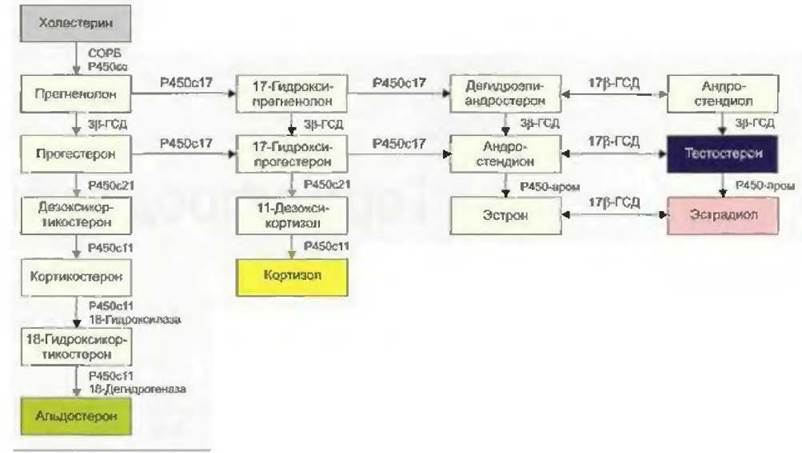
Рис. 7-1 Схема биосинтеза половых гормонов
Гонадный пол
В течение первых нескольких недель эмбрионального развития гонады обоего пола бывают индифферентными и бипотентными. Под влиянием TDF на 7-й неделе после зачатия зачатки гонад дифференцируются в яички. При отсутствии TDF (SRY) с 10-й недели начинается дифференцировка гонад в яичники.
Развитие яичников начинается на 10-й неделе после зачатия. На 14-й неделе в яичниках уже можно различить примордиальные фолликулы. До самого рождения яйцеклетки подвергаются редукционному делению, которое прерывается в профазе. Мейоз яйцеклетки завершается лишь непосредственно перед овуляцией. Большая часть овогоний, общее количество которых достигает 6—7 млн, к 20-й неделе атрезируется. К моменту рождения девочки у нее остается 1 млн овогоний, к моменту наступления менархе — 400 000. Лишь 400 яйцеклеток подвергаются овуляции (Baker, 1963).
Соматический пол
У эмбрионов вначале развиваются два парных половых протока, которые дают начало мужским или женским половым органам: вольфовы и мюллеровы протоки. При отсутствии яичек вольфовы протоки подвергаются обратному развитию, а из мюллеровых протоков развиваются матка, маточные трубы и проксимальный отдел влагалища. Наружные половые органы из индифферентных зачатков дифференцируются по женскому типу (урогенитальный синус, урогенитальный буорок), если не подвергаются вирилизирующему действию андрогенов. Наличия функционирующих яичников для этого не нужно, необходимо только отсутствие яичек.
В противоположность этому, дифференцировка по мужскому типу всегда представляет собой активный процесс. Яички плода продуцируют тестостерон и антимюллеров гормон. Под влиянием антимюллерова гормона, который вырабатывается клетками Сертоли, происходит обратное развитие мюллеровых протоков (Josso, 1975), в то время как высокая локальная концентрация стимулирует развитие вольфовых протоков (Siiteri u. Wilson, 1974). Из вольфовых протоков образуются придатки яичек, семявыносящие протоки и семенные пузырьки. Из устьев вольфовых протоков в урогенитальном синусе образуются простатическая и мембранозная части уретры.
Наружные половые органы в течение первых нескольких недель эмбрионального развития также индифферентны и бипотентны. При наличии яичек под влиянием тестостерона, вырабатываемого клетками Лейдига, происходит слияние уретральных и губно-мошоночных складок, из которых образуются пещеристые тела полового члена и мошонка. На 12-й неделе после зачатия развитие и образование мошонки завершается. Нарушение андрогенного влияния в течение этого периода всегда приводит к развитию половых органов по промежуточном) типу. После 12-й недели андрогены уже неспособны вызвать сращение губно-мошоночных складок, если оно к этому времени не было завершено.
Отсутствие у девочек яичек становится причиной обратного развития вольфовых протоков. Слияние мюллеровых протоков начинается с 6-й недели после зачатия. Из их краниального отдела образуются маточные трубы, каудальные отделы сливаются, образуя маточно-влагалищный канал. Этот зачаток связывается с эпителием урогенитального синуса, из которого формируется влагалище (O'Rahilly, 1977) (рис. 7-2).
Психический пол
Многочисленные эксперименты на животных показали, что как у низших млекопитающих, так и у приматов гормоны в критические фазы развития оказывают решающее влияние на циклическую (женскую) или тоническую (мужскую) секрецию гонадотропинов, на мужской и женский тип полового поведения и половой диморфизм развития головного мозга.
У людей знания добываются в процессе наблюдения за «экспериментами» самой природы. Большинство клинических исследований, касающихся гормональных нарушений в период развития, выполнены на пациентках с АГС. У них были описаны мужской тип поведения («девочки с мальчишеским озорством»), би- или гомосексуальная ориентация (Erhardt 1979; Erhardt u. Meyer/Bahlburg, 1981; Money et al., 1984), однако в исследованиях некоторых авторов эти данные не подтвердились (Muller et al., 1982).
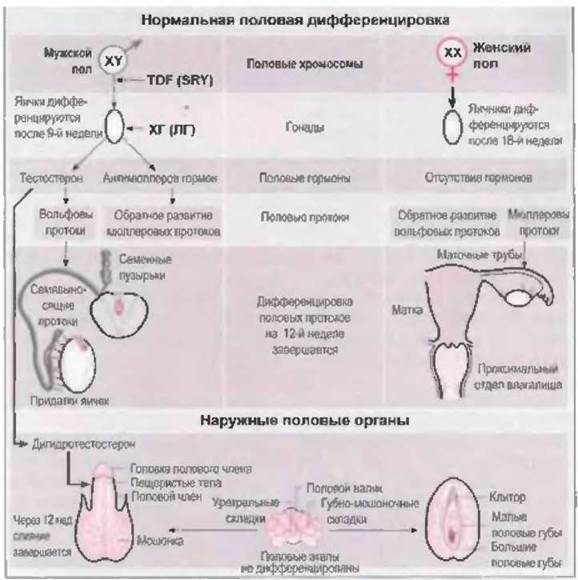
Рис. 7-2. Схема нормальной половой дифференцировки.
Девочки, матери которых во время беременности лечились гестагенными препаратами с андрогенным действием, обнаруживали мужской тип поведения (Money u. Erhardt, 1972). С другой стороны, гестагенные препараты, сходные по своему действию с прогестероном, препятствовали маскулинизирующему эффекте андрогенов и эстрогенов.
В отличие от АЕС, при полной резистентности к андрогенам (синдром тестикулярной феминизации) андрогенное влияние in utero отсутствует. Половая идентификация, а также тип поведения при этом женские (Masica et al., 1971). Уровень тестостерона в сыворотке крови этих пациенток соответствует верхней границе нормы для мужчин, и возможна беспрепятственная ароматизация его с превращением в эстрогены.
Пациентки с недостаточностью 5а-редуктазы рождаются с наружными половыми органами, развитыми по женскому типу и развиваются как девочки. В пубертатном периоде у них отмечается выраженная вирилизация. В группе таких пациенток из Доминиканской Республики в пубертатном периоде большинство изменили половую роль и половую идентичность (Imperato-McGinley et al., 1979). Мнение о том, что эти наблюдения подтверждают решающее значение биологических (гормональных) факторов в половой идентификации, оспаривается Rubin et al. (1981). Согласно этим авторам, в изменении половой идентичности пациенток, которые наблюдались Imperato-McGinley et al., решающую роль сыграли социокультурные условия в Доминиканской Республике, так как в сравнимой группе пациенток в США ни одна из них не перестала причислять себя к женскому полу, несмотря на вирилизацию в пубертатном периоде.
Из сказанного выше следует, что повышенный уровень андрогенов во внутриутробном периоде у пациенток с АЕС, несмотря на вирилизирующее действие на развитие половых органов, по-видимому, не оказывает существенного влияния на половую идентификацию и психосексуальную ориентацию (Erhardt, 1979; Erhardt et al., 1968; Money u. Schwarty, 1977). Половая идентификация, по- видимому, в значительной степени зависит от половой роли, в которой ребенок растет. Амбивалентная самоидентификация, вероятно, наблюдается в тех случаях, когда родители в воспитании ребенка не придерживаются строго определенной ролевой линии (Faiman u. Winter, 1974). Большинство исследователей основными в развитии половой идентификации считают либо врожденные, либо приобретенные факторы, в то время как речь идет скорее о взаимодействии гормональных и психосоциальных факторов (Rubin et al., 1981).
7.3. Нарушение половой дифференцировки
Нарушение половой дифференцировки может произойти на любой ее стадии. В зависимости от характера нарушения, степени его выраженности и стадии, на которой оно происходит, возможен широкий спектр патологических изменений — от незначительных отклонений, не имеющих клинического значения, до различной степени выраженности амбивалентности или полного изменения соматического пола по отношению к генетическому.
Истинный гермафродитизм, при котором у пациента имеются как яички, так и яичники, встречается редко. Чаще речь идет о пациентах с генетически детерминированным мужским полом, имеющих яички (и у которых при генетическом исследовании идентифицируют TDS), у которых маскулинизация недостаточно выражена (ложный мужской гермафродитизм), или о пациентках с генетически детерминированным женским полом, у которых, несмотря на функционирующие яичники, отмечается вирилизация (ложный женский гермафродитизм).
Ложный мужской гермафродитизм
При ложном мужском гермафродитизме пациенты имеют генетически детерминированный мужской пол (при генетическом исследовании у них выявляют TDS), яички, но недостаточно маскулинизированные внутренние и/или наружные половые органы.
Выраженность клинических проявлений широко варьирует — от гениталий, развитых полностью по женскому типу, и множества вариантов амбивалентных гениталий до внешне нормальных мужских гениталий с гипоспадией. Недостаточность вирилизации детей с генетически детерминированным мужским полом классифицируют в соответствии с данными, приведенными в табл. 7-1 и на рис. 7-3 (Sinnecker et al, 1985; Sinnecker et al., 1996).
Таблица 7-1. Классификация фенотипов при ложном мужском гермафродитизме (Sinnecker et al., 1997)
|
Тип |
Фенотип |
Клинические проявления |
|
1 |
Мужской |
Нарушение сперматогенеза и/или нарушение вирилизации в пубертатном периоде |
|
2 |
Преимущественно мужской |
Изолированная гипоспадия и/или микропенис Гипоспадия высокой степени, разделенная мошонка |
|
3 |
Амбивалентный |
Микропенис, напоминающий клитор. разделенная мошонка, напоминающая половые тубы, промежностно-мошоночная гипоспадия или урогенитальный синус с коротким, слепо заканчивающимся влагалищем |
|
4 |
Преимущественно женский |
Гипертрофия клитора и/или сращение половых туб, урогенитальный синус с коротким, слепо заканчивающимся влагалищем |
|
5 |
Женский |
Отсутствие признаков вирилизации до наступления пубертатного периода В пубертатном периоде: вирилизация, обусловленная недостаточностью 5а-редуктазы, феминизация, обусловленная дефектом андрогенных рецепторов |
Выделяют следующие причины недостаточной вирилизации:
• Функциональная недостаточность яичек у плода (дисгенезия гонад).
• Утрата какой-либо функции яичек, которые в остальном развиты нормально (дефект биосинтеза тестостерона, гипоплазия клеток Лейдига).
• Неспособность тканей реагировать на тестостерон. вырабатываемый нормально развитыми яичками (резистентность к андрогенным гормонам, недостаточность 5а-редуктазы).
Нарушения развития половых желез и хромосомные нарушения
Нарушение развития яичек бывает обусловлено в основном хромосомными аберрациями, однако в некоторых случаях причину его установить не удается. В зависимости от степени нарушения дифференцировки, а также от того, нарушено развитие одного или обоих яичек, половые органы индивидуума могут быть развиты по женскому типу, амбивалентно или по мужскому типу. В половине случаев в основе дисгенезии гонад лежит мозаицизм 45,X-/46,XY (Ferguson-Smith, 1965). Более чем у половины этих пациентов наружные половые органы развиты по женскому типу и имеются признаки вирилизации. Причиной этих отклонений считают структурные изменения Y-хромосомы в анафазе (Madan et al., 1979).
В отличие от отмеченной выше аномалии, при дисгенезии гонад у пациентов с кариотипом 46,XY структурные изменения в Y-хромосоме отсутствуют. Этиология некоторых заболеваний, проявляющихся различными аномалиями развития, установлена: мутации гена WT1 обнаруживаются при синдромах Дени—Драша и WAGR (Mueller, 1949), при синдроме Смита—Лемли—Опица отмечается недостаточность дегидрохолестерин-С7- редуктазы, которая является причиной повышения уровня 7-дегидрохолестерина в сыворотке крови (Tint et al., 1995). Дисгенезию гонад может вызвать также удвоение локуса DSS на Хр21 и мутации гена SOX9 на 17q21, в этих случаях она сочетается с кампомелической дисплазией (Wagner et al., 1949).
Неполная (смешанная) дисгенезия гонад
Изменения в яичках при нарушении их дифференцировки могут быть выражены в различной степени, одинаково с обеих сторон или быть асимметричными. Они могут затрагивать одно яичко при нормально развитом друом, оба яичка в различной степени вплоть до полной дисгенезии обоих яичек, которые при этом имеют вид соединительнотканных тяжей, расположенных в позиции яичников (строковые гонады). Такое разнообразие морфологических вариантов обусловливает различную степень функциональных нарушений. Недостаточная секреция тестостерона и антимюллерова гормона приводит к недостаточной стимуляции вольфова протока на стороне поражения и гипоплазии семенных пузырьков, семявыносящего протока и придатка яичка. Недостаточная секреция также нарушает обратное развитие мюллерова протока на стороне поражения, что обусловливает персистенцию образующихся из него анатомических структур (маточная труба, матка, проксимальный отдел влагалища) с различной степенью их дисплазии (рис. 7-4).
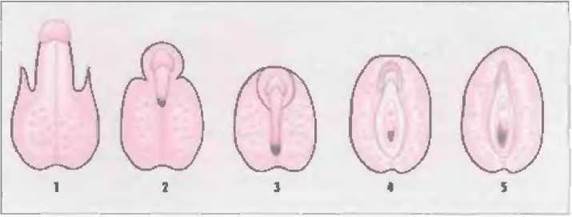
Рис. 7-3. Классификация фенотипов при ложном мужском гермафродитизме (Sinnecker et al„ 1996,1997).
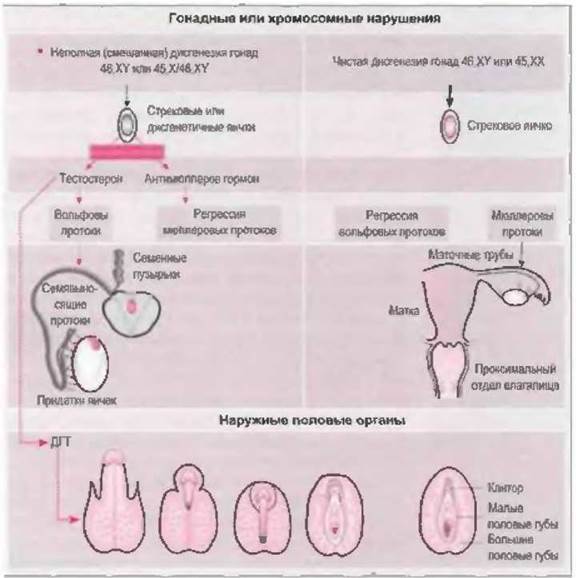
Рис. 7-4. Неполная дисгенезия гонад (пораженная область при тех или иных нарушениях окрашена в красный цвет или затушевана)
Клиническая картина
Клиническая картина охватывает женский, амбивалентный и мужской фенотипы и все многообразие промежуточных форм. Потеря функции дисгенетичным яичком обычно одинаково влияет на секрецию тестостерона и антимюллерова гормона. Если секреция нарушена настолько, что приводит к недостаточной маскулинизации наружных половых органов и развитию их по промежуточному или женскому типу, то обычно наблюдается персистенция анатомических структур, образующихся из мюллеровых протоков (маточные трубы, матка, проксимальный отдел влагалища). Эта важная особенность дисгенезии гонад, клинические проявления которой столь разнообразны, позволяет отличить ее от других причин ложного мужского гермафродитизма (рис. 7-5).
Диагностика
Заподозрить неполную дисгенезию гонад следует в тех случаях, когда у пациента с гениталиями промежуточного типа имеется матка и либо пальпируются гонады (это всегда яички), либо при генетическом анализе обнаруживают Y-хромосому.

Кариотип обычно бывает 46,XY, 45,X/46,XY, 45,X/47,XYY или подобный этому. Наружные половые органы развиты по женскому (отмечается гипертрофия клитора), промежуточному или мужскому типу. Отмечается различная степень дифференцировки вольфовых протоков и соответственно обратного развития мюллеровых протоков. Гонады представлены соединительнотканными тяжами (стрековые) либо дисгенетичные или одно яичко дисгенетично, а друое развито нормально (смешанная дисгенезия гонад). Половое созревание не наступает или отмечается вирилизация. Если появляется гинекомастия, ее причиной обычно бывает гонадобластома, продуцирующая эстрадиол. Концентрация гонадотропинов (ЛГ, ФСГ) в плазме крови со 2-го года жизни повышена, тестостерона — снижена.
Лечение
При решении вопроса, какой пол следует выбрать для растущего ребенка, исходят из того, при каком поле скорее всего следует рассчитывать на нормальную функцию наружных половых органов. Это определяют в зависимости от анатомических особенностей, возможности хирургической коррекции и культурного уровня семейного окружения ребенка.
Если ребенок будет воспитываться как девочка, яички следует удалить. Оставление их таит в себе двоякую опасность. Во-первых, остаточная функция яичек обусловит гетеросексуальное (мужское) течение пубертатного периода. Во-вторых, дисгенетичные гонады, клетки которых содержат Y-хромосому, часто малигнизирутотся. Такая предрасположенность к развитию гонадобластомы объясняется присутствием на Y-хромосоме в так называемом гонадобластомном локусе гена GBY (Tsuchiya, 1995). Риск злокачественного перерождения в 10-летнем возрасте составляет 2%, в 20- летнем — 16%, в 30-летнем — 27,5% (Manuel et al., 1976). Обычно развивается гонадобластома или герминома. В период, соответствующий пубертатному", детям проводят заместительнуто терапию эстрогенами и гестагенами.
Полная (чистая) дисгенезия гонад
При полной, или чистой, дисгенезии гонад отмечается фиброзное их перерождение (стрековые гонады). Дети имеют кариотип 46,XY или 46,ХХ. Дисгенезия гонад при кариотипе 46,XY в 10% случаев бывает обусловлена мутацией гена SRY (Ferguson-Smith u. Goodfellow, 1995). В остальных случаях причиной дисгенезии, по-видимому, бывают мутации аутосомных генов дифференцировки.
Клиническая картина
При полной дисгенезии гонад отмечается нормальный женский фенотип (внутренние и наружные половые органы развиты нормально, по женскому" типу). Схематически это показано на рис. 7-6. У большинства этих пациенток отсутствуют признаки полового созревания в пубертатном периоде и отмечается первичная аменорея. У пациенток с кариотипом 46.XY (синдром Суайера) чаще наблюдается гипертрофия клитора. Риск злокачественного перерождения стрековых гонад, клетки которых содержат Y-хромосому, такой же, как при смешанной дисгенезии гонад. Однако злокачественное перерождение дисгенетичных гонад, не содержащих Y-хромосому, является редкостью (см. рис. 7-6 и 7-7).
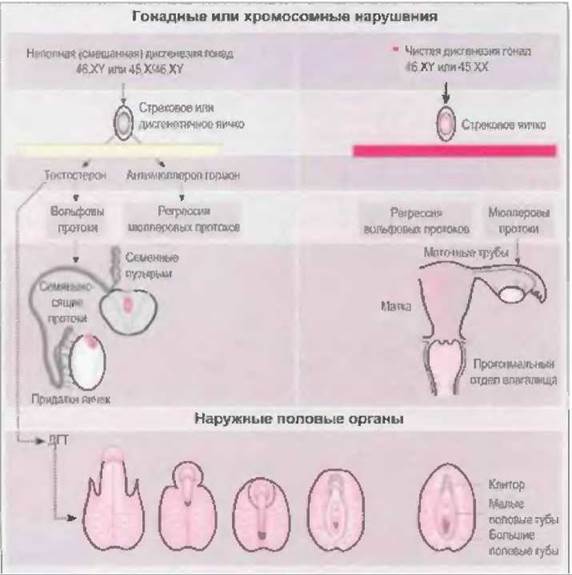
Рис. 7-6. Полная дисгенезия гонад (пораженная область при тех или иных нарушениях окрашена в красный цвет или затушевана)
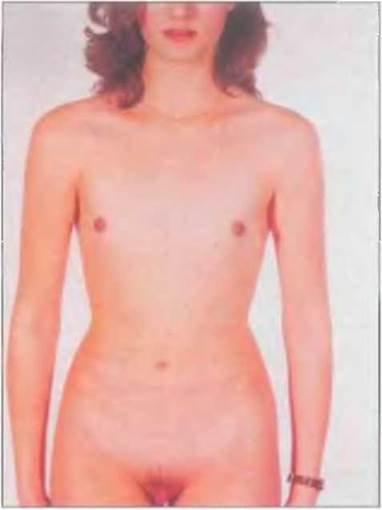
Рис. 7-7. Пациентка с дисгенезиеи гонад и кариотипом 46.XY (Sinnecker et ot 1982).
Диагностика
Диагноз полной дисгенезии гонад ставят на основании отсутствия признаков полового созревания и гипогонадотропного гипогонадизма у пациентки с нормальным мужским или женским набором хромосом. При УЗИ яичники у них не идентифицируются, в остальном наружные и внутренние половые органы развиты нормально, по женскому типу.
Лечение
Лечение заключается в заместительной терапии эстрогенами и гестагенами, назначаемой в возрасте, соответствующем пубертатному периоду. В связи с высоким риском злокачественного перерождения стрековые гонады у пациенток с кариотипом 46.XY следует удалить в ранние сроки (Sinnecker et al., 1982). Механизмы развития ложного мужского гермафродитизма приведены на рис. 7-8.
Нарушение биосинтеза тестостерона
Этиология и патогенез
О нарушении биосинтеза тестостерона свидетельствует недостаточная вирилизация у лиц с генетически мужским полом и выраженным в различной степени женским фенотипом. Поскольку выработка антимюллерова гормона не нарушена, то, в отличие от дисгенезии гонад, происходит полная регрессия мюллеровых протоков. Нарушение следующих трех звеньев может вызвать нарушение биосинтеза глюкокортикоидных и половых гормонов:
• Недостаточность СОРБ.
• Недостаточность Зв-гидросистероиддегидрогеназы типа 2.
• Недостаточность СУР17-(17а-гидроксилазы-17,20-лиазы).
• Недостаточность следующих двух ферментов вызывает нарушение биосинтеза только половых гормонов: СУР17-(17,20-лиазы).
• 17(3 -гидроксистероиддегидрогеназы типа 3.

Рис. 7-8. Механизмы развития ложного мужского гермафродитизма (пораженная область при тех или иных нарушениях окрашена в красный цвет или затушевана).
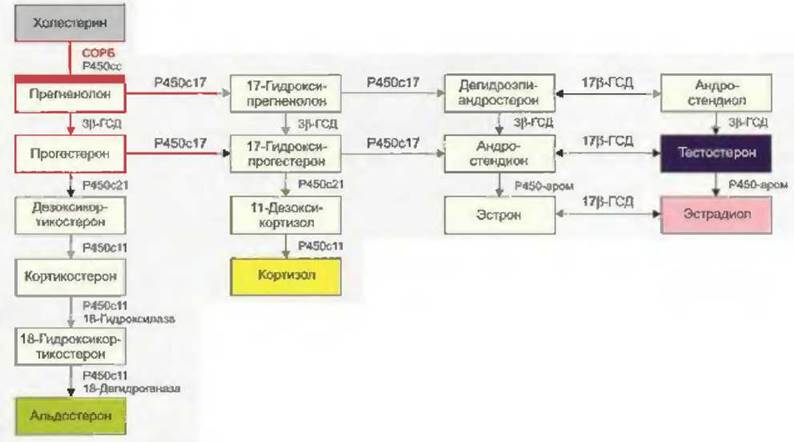
Рис. 7-9. Схема биосинтеза стероидных гормонов при недостаточности СОРБ.
Недостаточность СОРБ
Холестерин переносится с помощью СОРБ с наружной поверхности митохондриальной мембраны на внутреннюю, где при участии фермента CYP11A1 (цитохром P450scc) используется в качестве субстрата для биосинтеза стероидных гормонов (Bose et al., 1996). Недостаточность СОРБ вызывает нарушение синтеза всех стероидных гормонов в равной мере. Клинически это проявляется симптомами недостаточности глюко- и минералокортикоидов и половых гормонов.
Клиническая картина
Ребенок имеет женский или амбивалентный фенотип. Влагалище заканчивается слепо, производные мюллеровых протоков отсутствуют. Вольфовы протоки находятся в рудиментарном состоянии. Тяжелая недостаточность коркового вещества надпочечников, если ее не диагностировать и не лечить своевременно, быстро приводит к смерти. Все выжившие к настоящему времени дети росли как девочки. В пубертатном периоде возможны легкие проявления вирилизации (рис. 7-9 и 7-10).
Диагностика
Диагностическое значение имеют очень низкий уровень всех стероидных гормонов в плазме крови (глюкокортикоидов, минералокортикоидов, половых гормонов) и повышенный уровень АКТГ, ренина, иногда — гонадотропинов, гиперплазия надпочечников со смещением почек в каудальном направлении при УЗИ и ги- перпигментированная кожа, имеющая бронзовый цвет. Стимуляция АКТГ не вызывает повышения уровня кортизола в плазме крови. Диагностика должна включать также анализ ДНК.
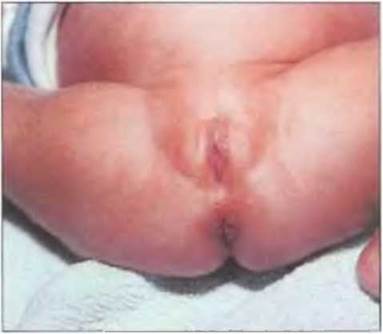
Рис. 7-10. Пациентка (кариотип 46,XY) с нарушением биосинтеза стероидов в надпочечниках и половых железах. Обращает на себя внимание бронзовый цвет кожи.
Лечение
Жизненно необходима заместительная терапия минерало- и глюкокортикоидами. Во избежание вирилизации в пубертатном периоде гонады желательно удалить до его наступления. В пубертатном периоде назначают эстрогены. Для индуцирования роста волос на лобке назначают малые дозы тестостерона.
Недостаточность 3b- гидрокеистероиддетидрогеназы
Этиология и патогенез
При недостаточности 3р-гидроксистероидде- гидрогеназы происходит блокирование биосинтеза стероидных гормонов на ранней его стадии, в результате возникает недостаточность как глюко- и минералокортикоидов, так и половых гормонов. В основе заболевания лежит мутация гена HSD3B2, кодирующего фермент 3р-гидроксисте- роиддегидрогеназа-А4 5-изомеразу типа 2, который находится в основном в надпочечниках и половых железах.
Клиническая картина
Клинические проявления отличаются большим разнообразием. Отмечаются амбивалентные гениталии с микропенисом, гипоспадия, неполное сращение губно-мошоночных складок, персистенция урогенитального синуса, слепо заканчивающееся влагалище и в зависимости от степени недостаточности фермента — выраженные в различной степени симптомы недостаточности коркового вещества надпочечников. Внутренние половые органы развиты нормально, по мужскому тишу производные мюллеровых протоков отсутствуют. В пубертатном периоде отмечаются незначительная вирилизация и гинекомастия.
Диагностика
Для заболевания характерна повышенная концентрация 17р-гидроксипрегненолона и де гидроэпиандростерона. Поскольку концентрация этих гормонов может быть повышенной и у здоровых новорожденных, целесообразно провести стимуляционную пробу с АКТГ и ХГ. Диагностика включает также анализ ДНК.
Лечение
Назначают заместительную терапию глюкокортикоидами, а также минералокортикоидами (в зависимости от степени недостаточности Зв- гидроксистероиддегидрогеназы). В пубертатном периоде проводят заместительную терапию половыми гормонами. При микропенисе начинают раннюю терапию тестостероном (Chung et al., 1987).
Недостаточноть фермента СУР -(17 а- гидроксилазы -17,20 -лиазы)
Фермент CYP (цитохром Р450с17) катализирует гидроксилирование прегненолона и прогестерона в 17а положении (гидроксилазная активность) и отщепление боковой цепи от образовавшегося 17 а- гидроксилированного субстрата (17,20-лиазная активность). Несмотря на то что этот фермент кодируется одним геном (Conte et al., 1994), активность его может нарушаться по-разному. Так, возможно нарушение одновременно гидроксилазной и 17,20- лиазной активности или изолированное нарушение одной из них. В зависимости от этого клиническая картина проявляется симптомами недостаточности 17а-гидроксилазы-17,20-лиазы или изолированной недостаточности 17,20-лиазной активности.
Недостаточность 17а-гидроксилазной активности приводит к нарушению синтеза глюкокортикоидов и половых гормонов. Повышенное образование 11-дезоксикортикостерона и кортикостерона вызывает артериальную гипертензию, гиперкалиемию и подавление активности ренина плазмы. Недостаточность бывает выражена в различной степени, что и определяет клинические проявления.
Клиническая картина
Для заболевания характерно разнообразие клинических вариантов. Больные могут иметь генетически детерминированный мужской пол при женском фенотипе со слепо заканчивающимся влагалищем, различные варианты амбивалентных половых органов или мужской фенотип в сочетании с микропенисом и гипоспадией. Производные вольфовых протоков гипопластичны, производные мюллеровых протоков отсутствуют. В пубертатном периоде возможны вирилизация и гинекомастия. Минералокортикоидный эффект высоких концентраций дезоксикортикостерона и кортикостерона вызывает задержку натрия и воды, потерю калия, увеличение объема циркулирующей плазмы, артериальную гипертензию с гипоренинемией и алкалозом.
Диагностика
Характерна повышенная концентрация в плазме АКТГ, 11 -дезоксикортикостерона, кортикостерона и прогестерона и пониженная концентрация альдостерона, 17а-гидроксипрогестерона, кортизола и половых гормонов. Отмечаются также гипоренинемическая артериальная гипертензия и гипокалиемический алкалоз.
Лечение
Заместительная терапия глюкокортикоидами приводит к нормализации артериального давления и уровня калия в сыворотке крови. Половые гормоны назначают в пубертатном периоде. Из-за возможности вирилизации у лиц с генетически детерминированным мужским полом до наступления пубертатного периода яички желательно удалить.
Изолированная недостаточность CYP-(17,20- лиазы)
Недостаточность 17.20-лиазной активности фермента CYP17 (цитохром Р450с17) приводит к нарушению превращения 17а-гидроксипрег- ненолона и 17а-гидроксипрогестерона в андрогенные гормоны дегидроэпиандростерон и андростендион.
Клиническая картина
Фенотип лиц с генетически детерминированным мужским полом может быть женским со слепо заканчивающимся влагалищем, амбивалентным или преимущественно мужским. Вольфовы протоки дифференцированы нормально или гипопластичны, производные мюллеровых протоков отсутствуют. В пубертатном периоде отсутствуют вторичные половые признаки или появляются легкие симптомы вирилизации.
Диагностика
Диагностическое значение имеют повышенная концентрация C2i -стероидов 17а-гидроксип- рогестерона и 17а-гидроксипрегненолона, сниженная концентрация Сі9-стероидов дегидроэпиандростерона и андростендиона, а также тестостерона и эстрадиола. При стимуляции АКТГ или ХГ отношение С21-стероиді/С19-стероиді значительно повышается.
Лечение
Заместительную терапию половыми гормонами проводят в пубертатном периоде. При генетически детерминированном мужском поле у пациента, который рос и воспитывался как девочка, до наступления пубертатного периода яички следует удалить, чтобы избежать вирилизации.
Недостаточность 17b-гидроксистероиддегидрогеназы
Из 5 изоферментов 17в-ГСД наибольшее значение ДЛЯ дифференцировки ПО мужскому типу имеет экспрессирующийся в яичках фермент 17в- ГСД типа 3, который катализирует последнюю ступень биосинтеза половых гормонов. На этой ступени происходит превращение дегидроэпи- андростерона в А5-андростендиол, андростендиона в тестостерон и эстрона в 17в-эстрадиол. Недостаточность тестостерона приводит к полному отсутствию маскулинизации у плода мужского пола во внутриутробном периоде. Однако в пубертатном периоде в результате экстрагландулярного превращения имеющегося в избытке андростендиона в тестостерон другими изоферментами 17р-ГСД происходит выраженная вирилизация.
Клиническая картина
В большинстве случаев лица с генетически детерминированным мужским полом имеют женские наружные половые органы со слепо заканчивающимся влагалищем, иногда отмечаются легкие симптомы вирилизации. Вольфовы протоки дифференцированы, производные мюллеровых протоков отсутствуют. В пубертатном периоде происходит выраженная вирилизация с ломкой голоса и увеличением клитора. Иногда отмечается гинекомастия. Как и при недостаточности 5а- редуктазы, описаны случаи изменения женской половой роли на мужскую при начавшейся вирилизации.
Диагностика
Начиная с пубертатного периода повышается концентрация андростендиона и эстрона в плазме крови, концентрация тестостерона и эстрадиола снижается. В препубертатном периоде необходима стимуляция ХГ, она приводит к выраженному увеличению отношения андростендион/ тестостерон и эстрон/эстрадиол. Диагностика включает в себя также анализ ДНК, в частности rcHaHSD17B3.
Лечение
У лиц с генетически детерминированным мужским полом, которые росли и воспитывались как девочки, следует удалить яички до наступления пубертатного периода. При амбивалентных половых органах может встать вопрос о мужской половой ориентации. В таких случаях в детском возрасте проводят терапию тестостероном для увеличения размеров полового члена, которую дополняют хирургической коррекцией, направленной на маскулинизацию половых органов. В пубертатном периоде следует назначить заместительную терапию тестостероном для достижения полной маскулинизации и предупреждения гинекомастии (рис. 7-11 и 7-12).
Гипоплазия клеток Лейдига
Редкой причиной ложного мужского гермафродитизма является отсутствие чувствительности клеток Лейдига к ХГ и ЛГ (агенезия или гипоплазия клеток Лейдига).
Этиология и патогенез
Гипоплазия или агенезия клеток Лейдига объясняется недостаточностью рецепторов, которая делает эти клетки нечувствительными к влиянию ХГ и ЛГ (Themmen u. Вгаппег, 1996). Недостаточная выработка тестостерона приводит к недостаточной маскулинизации плода мужского пола во внутриутробном периоде. Поскольку, в отличие от дисгенезии гонад, клетки Сертоли сохранены и нормально функционируют, т. е. продуцируют антимюллеров гормон, то у лиц с этой патологией производные мюллеровых протоков отсутствуют. У некоторых пациентов вольфовы протоки бывают дифференцированы, несмотря на недостаточную маскулинизацию наружных половых органов. Это, по-видимому, объясняется паракринной секрецией тестостерона, которого достаточно для локального действия: индуцирования дифференцировки вольфовых протоков. Однако концентрация тестостерона в плазме остается слишком низкой, чтобы вызвать достаточную маскулинизацию наружных половых органов. Клинические проявления недостаточности тестостерона соответствуют характеру нарушений его биосинтеза.
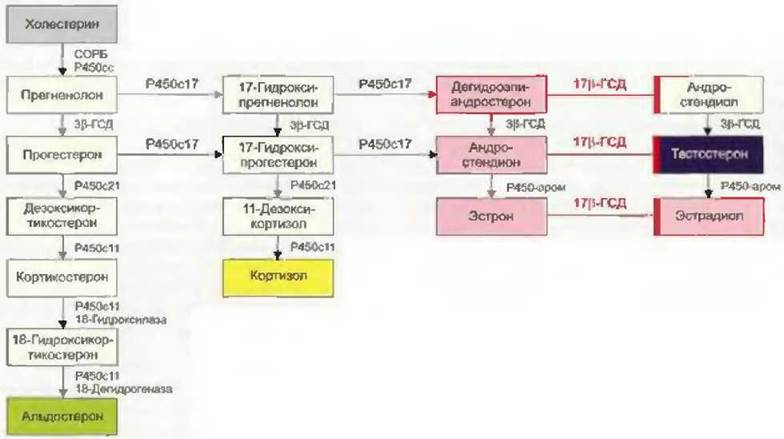
Рис. 7-11. Схема нарушения биосинтеза стероидных гормонов при недостаточности 17(3 -ГСД.
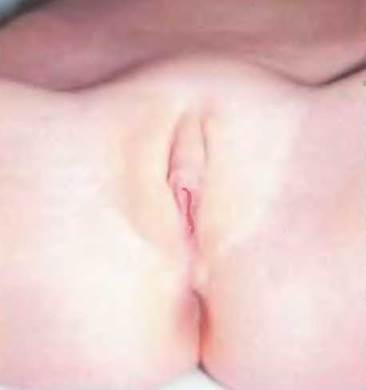
Рис. 7-12. Пациентка с недостаточностью 17(3 -ГСД.
Клиническая картина
Наружные половые органы обычно развиты по женскому типу со слепо заканчивающимся влагалищем и легкими признаками вирилизации, но могут быть также амбивалентными или развиты по мужскому типу. Отмечаются развитые в различной степени производные вольфовых протоков, производные мюллеровых протоков отсутствуют. Признаки полового созревания в пубертатном периоде не появляются.
Диагностика
Уровень тестостерона в плазме низкий, однако в препубертатном периоде он не имеет диагностического значения. После стимуляции ХГ происходит небольшое повышение уровня тестостерона, но без повышения уровня его предшественников, которое наблюдается при нарушении его биосинтеза. Связывание ХГ и ЛГ с рецепторами клеток Лейдига незначительное.
Лечение
Выбор пола определяется по степени маскулинизации. Заместительную терапию половыми гормонами проводят ко времени наступления пубертатного периода.
Недостаточность стероид-5а-редуктазы типа 2
Новаковский и Ленц в 1961 г. впервые описали форму ложного гермафродитизма, наследуемую по аутосомно-рецессивному типу. Они назвали ее псевдовагинальной перинеоскротальной гипоспадией (Nowakowski u. Lenz, 1961). В дальнейшем Императо-Магинли и соавт. (Imperato-McGinley et al., 1974) показали, что в основе этого заболевания лежит недостаточность стсроид-5а-рсдуктазы типа 2, кодируемой геном SRD5A2.
Этиология и патогенез
Превращение тестостерона в биологически более активный дигидротестостерон происходит при участии фермента стсроид-5а-рсдуктазы типа 2, кодируемого геном SRD5A2 (Andersson et al., 1991). Известны различные дефекты этого фермента. Чаще речь идет об уменьшении сродства к тестостерону и тем самым снижении активности фермента (Moore et al., 1975). К друим дефектам относятся изменения, приводящие к уменьшению стабильности фермента и его сродства к кофактору NADPH (Leshin et al., 1978). Во всех случаях нарушается превращение тестостерона в дигидротестостерон в тканях. Это проявляется в недостаточной маскулинизации наружных половых органов. Развитие вольфовых протоков, находящееся исключительно под влиянием паракриннои секреции тестостерона, а также регрессия мюллеровых протоков не нарушены. В пубертатном периоде происходит выраженная вирилизация, несмотря на сохраняющуюся недостаточность 5ос-редуктазы. Секреция эстрадиола нормальная, гинекомастия не наблюдается.
Клиническая картина
У детей с генетически детерминированным мужским полом недостаточность стероид-5а-редуктазы типа 2 вызывает выраженное в различной степени нарушение маскулинизации. Клинически это может проявиться внешне нормальными наружными половыми органами, развитыми по мужскому типу, различными вариантами амбивалентных половых органов, а также полностью женским фенотипом. Яички бывают расположены в паховых каналах, губномошоночных складках или мошонке. Производные вольфовых протоков полностью развиты (придатки яичек, семявыносящие протоки и семенные пузырьки). Семявыносящие протоки иногда открываются во влагалище. Производные мюллеровых протоков отсутствуют (Sinnecker et al., 1996; Walsh, 1974).
В пубертатном периоде на фоне нормального повышения уровня тестостерона отмечается выраженная вирилизация с ломкой голоса и увеличением размеров полового члена без появления гинекомастии.
Диагностика
В детском возрасте определение концентрации гормонов в сыворотке крови диагностического значения не имеет. После стимуляции ХГ отмечается существенное повышение отношения тестостерон/дигидротестостерон (Hiori et al., 1996). Снижение активности фермента можно выявить и в культуре фибробластов, полученных из кожи половых органов (Pinsky et al., 1978). Анализ гена SRD5A2 позволяет подтвердить диагноз даже при минимальной недостаточности фермента (Hiori et al., 1996: Sinnecker et al., 1996).
Лечение
Дети, y которых половой член выражен, несмотря на малые размеры, должны воспитываться как мальчики. Раннее лечение дигидротестостероном способствует увеличению размеров полового члена и облегчает хирургическую коррекцию гипоспадии. После пубертатного периода также показана терапия высокими дозами тестостерона для усиления вирилизации (Price et al., 1984).
У детей, которых воспитывают как девочек, до наступления пубертатного периода следует обязательно удалить яички во избежание вирилизации. В пубертатном периоде проводят заместительную терапию эстрогенными и гестагенными препаратами (рис. 7-13 и 7-14).
Синдром резистентности к андрогенам
Резистентность к андрогенам является второй после дисгенезии гонад причиной ложного мужского гермафродитизма. Несмотря на нормальную или даже повышенную концентрацию андрогенов в плазме крови, отмечается недостаточность андрогенного влияния на ткани. Клинически синдром может проявиться полностью женским фенотипом (тип 5, см. рис. 7-3 и табл. 7-1) при полной резистентности к андрогенам (синдром тестикулярной феминизации), различными вариантами амбивалентных гениталий (типы 4—2) или мужским фенотипом, при котором у пациентов отмечаются частичная резистентность к андрогенам и бесплодие (тип I).
Этиология и патогенез
Причиной резистентности к андрогенам является недостаточность андрогенных рецепторов.
Она может быть обусловлена нарушением связывания андрогенов с рецепторами, транслокацией комплекса стероидный гормон—рецептор из цитоплазмы в клеточное ядро, димеризацией рецепторов и связыванием их с ДНК и последующей транскрипцией и трансляцией.
Белок андрогенного рецептора кодируется геном, локализованным на длинном плече Х-хромосомы (Xqll-12). Этот белок в С-концевой области имеет домены, связывающие ДНК и гормон (рис. 7-15).
Почти во всех случаях резистентности к андрогенам удается выявить мутации гена, кодирующего андрогенный рецептор. Чаще это точковые мутации, затрагивающие одну аминокислоту. В зависимости от локализации мутации рецепторная недостаточность проявляется по-разному — от полной утраты активности рецептором (Burstein et al., 1979; Marcelli et al., 1990) до изменения качества связи с гормоном (McPhaul et al., 1991) или нарушения связывания комплекса стероидный гормон—рецептор с ДНК. Замена одного или нескольких оснований кодирующей последовательности гена при нарушенном сплайсинге может привести к преждевременному появлению стоп-кодона и утрате рецептором ДНК-и гормонсвязывающего доменов (Hiort et al., 1996; Marcelli et al., 1990; McPhaul et al., 1991). Помимо точковых мутаций, возможны также делеции и инсерции, которые, как правило, вызывают полную резистентность к андрогенам. Обычно пациенты с одинаковой мутацией имеют сходный фенотип, хотя в отдельных случаях фенотипы у больных членов одной семьи могут сильно различаться (рис. 7-16, а и б).
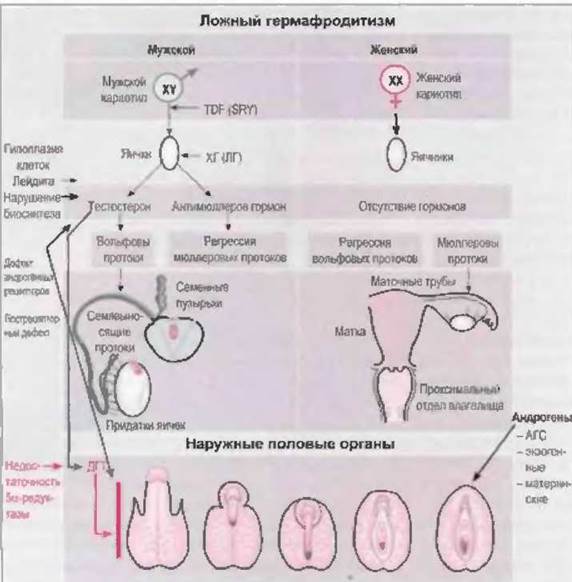
Рис. 7-13. Недостаточность стероид-5а- редуктазы типа 2.
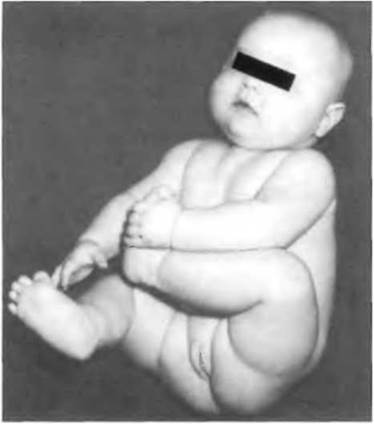
Рис. 7-14. Пациентка (кариотип 46.XY) с недостаточностью 5а- редуктазы (Sinnecker et al„ 1996).
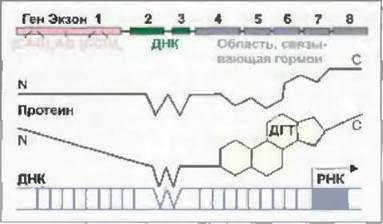
Рис. 7-15. Схематическое изображение гена белка андрогенного рецептора и взаимодействия активированного комплекса андрогенный рецептор-дигидротестостерон с ДНК.
Полная резистентность к андрогенам
Если действие андрогенов полностью блокируется, в эмбриональном периоде происходит развитие вольфовых протоков или маскулинизация наружных половых органов. Поскольку выработка антимюллерова гормона и его действие не нарушаются, происходит полная регрессия мюллеровых протоков. У детей концентрация половых гормонов и гонадотропинов в сыворотке крови низкая и соответствует возрастной норме (Fakhiy et al., 1988). В пубертатном периоде в гипоталамо-гипофизарной системе, которая также резистентна к андрогенам, увеличивается амплитуда и частота пульсирующей секреции ЛГ (Brown et al., 1990), что вызывает повышеннуто выработку яичками тестостерона (Tsuchiya et al., 1995) и эстрадиола (MacDonald et al., 1979). В результате экстрагландулярной ароматизации накапливающегося в крови тестостерона происходит дальнейшее увеличение концентрации эстрадиола, которое вызывает выраженную феминизацию.
Клиническая картина
Клиническая картина при полной резистентности к андрогенам характеризуется женским фенотипом (см. рис. 7-16, а) при генетически детерминированном мужском поле и хорошо развитыми женскими вторичными половыми признаками, появляющимися в пубертатном периоде (тип 5). Вторичное оволосение обычно слабо выражено. Клитор нормальных размеров, малые половые губы недостаточно развиты, влагалище слепо заканчивается. Производные мюллеровых (матка, маточные трубы) и вольфовых протоков отсутствуют. Глубина влагалища обычно достаточна для нормальной половой жизни. Половая идентификация женская. Частота полной резистентности к андрогенам составляет I на 20 000 рождений мальчиков (Grumbach u. Conte, 1998). У детей иногда обнаруживают яички в паховых каналах или паховые грыжи. Однако основанием для диагностики синдрома часто становится отсутствие менструаций, несмотря на появление признаков полового созревания.
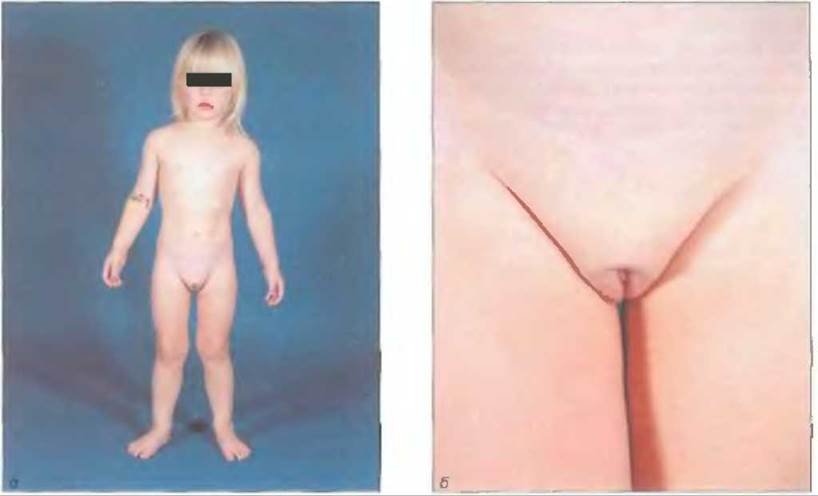
Рис. 7-16. Полная резистентность к андрогенам, о-пациентка (кариотип 46. XY ) с полной резистентностью к андрогенам типа 5; б- наружные половые органы этой пациентки.
Диагностика
Характерные клиническая картина и гормональный профиль — феминизация, первичная аменорея, слепо заканчивающееся влагалище, повышенный уровень ЛГ и резко повышенный уровень тестостерона в сыворотке крови в постпубертатном периоде — позволяют поставить правильный диагноз.
До наступления пубертатного периода клиническая картина и гормональный профиль неинформативны. Заподозрить синдром позволяют расположенные в паховых областях или в половых губах яички. Иногда яичко обнаруживают во время операции, выполняемой по поводу паховой грыжи. В таких случаях для подтверждения диагноза проводят пробу на резистентность к андрогенам и молекулярно-генетический анализ, позволяющий выявить мутации в гене рецептора к андрогенам.
Проба на резистентность к андрогенам основана на биологическом эффекте анаболического андрогенного гормона станозолола на концентрацию СССГ в сыворотке крови. В норме после введения 0,2 мг/кг станозолола в течение 3 дней концентрация СССГ в сыворотке крови через 5—8 дней снижается наполовину. У пациентов с полной резистентностью к андрогенам (тип 5) такого снижения не происходит, а при частичной резистентности к андрогенам концентрация СССГ снижается в различной степени в зависимости от недостаточности маскулинизации, но это снижение выражено в меньшей степени, чем в норме (Sinnecker u. Kohler, 1989; Sinnecker et al., 1997). Результаты пробы на резистентность к андрогенам приведены на рис. 7-17. Эта проба позволяет оценить степень резистентности к андрогенам и тем самым дополняет информацию, получаемую при структурном анализе ДНК.
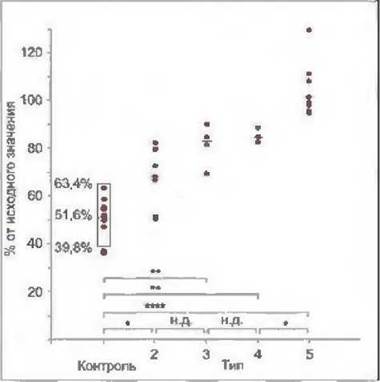
Рис. 7-17. Проба на резистентность к андрогенам (Sinnecker et al„ 1997). Снижение концентрации СССГ в сыворотке крови через 3 дня после введения станозолола (0,2 мг/кг) выражено в процентах от исходного значения у пациентов с резистентностью к андрогенам (типы 2-5) и у пациентов контрольной группы с различными аномалиями развития половых органов, но нормальной чувствительностью к андрогенам.
Точками обозначены пациенты, линиями - медианы в каждой группе, Звездочки обозначают степень достоверности различий между исследованными группами пациентов (н,д, - недостоверно; * - <0,05; ** - <0,01 ; *** - <0,001 ; **** - <0,0001 ).
Лечение
Лечение полной резистентности к андрогенам не представляет трудностей. Яички следует по возможности сохранить до завершения пубертатного периода, чтобы обеспечить спонтанное развитие по женскому типу. После пубертатного периода их необходимо удалить, так как риск злокачественного их перерождения на третьем десятилетии жизни возрастает до 4—10% (Manuel et al., 1976; Rutgers u. Scully, 1991; Verp u. Simpson, 1987). После гонадэктомии назначают заместительную терапию эстрогенами и гестагенами. При неглубоком влагалище его улубляют с помощью экспандера.
Частичная резистентность к андрогенам
Если биологическое действие андрогенов нарушено частично, в эмбриональном периоде стимуляция вольфовых протоков и маскулинизация наружных половых органов оказываются неполными. Поскольку выработка антимюллерова гормона и его эффект не нарушены, то происходит полная регрессия мюллеровых протоков.
Клиническая картина
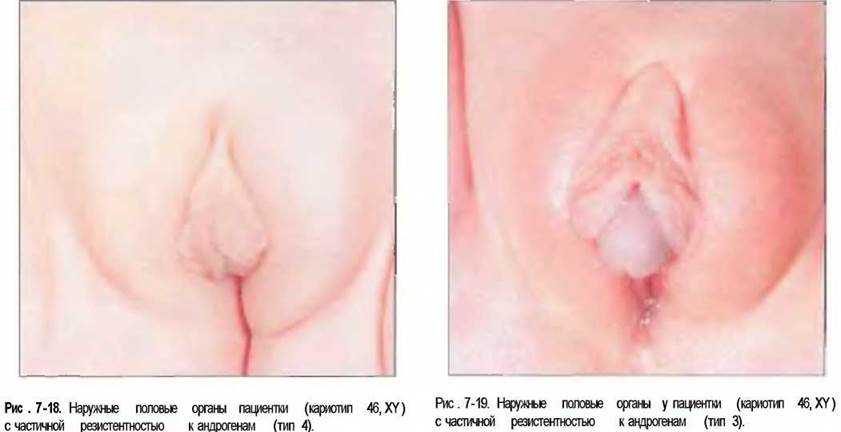
В зависимости от степени недостаточности рецепторов андрогенов пациенты имеют преимущественно женский фенотип, слепо заканчивающееся влагалище и легкие признаки вирилизации (тип 4), различные варианты амбивалентных гениталий (типы 3 и 2) или нормальный мужской фенотип, при котором, однако, страдают бесплодием, обусловленным азооспермией (тип 1). Фотографии половых органов таких пациентов приведены на рис. 7-18—7-20. В пубертатном периоде из-за частичного эффекта андрогенов, несмотря на высокую концентрацию тестостерона в сыворотке крови, мужские вторичные половые признаки выражены относительно слабо. С другой стороны, повышенная экстрагландулярная продукция эстрогенов приводит к развитию гинекомастии.
Разнообразие клинических форм заболевания велико. Хотя, учитывая Х-сцепленный рецессивный тип наследования, в одной и той же семье пациенты должны иметь одинаковый генетический дефект рецептора, степень маскулинизации у них может быть различной. Разнообразие клинических проявлений требует проведения дифференциальной диагностики с другими формами ложного мужского гермафродитизма.
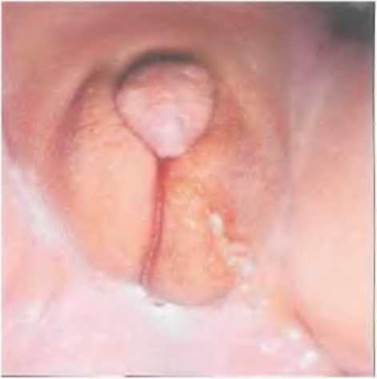
Рис. 7-20. Наружные половые органы пациента (кариотип 46, XY) с частичной резистентностью к андрогенам (тип 2),
Диагностика
Развитие наружных половых органов отличается значительной вариабельностью. Вольфовы протоки у пациентов либо отсутствуют, либо гипопластичны или развиты нормально. Производных мюллеровых протоков обычно не бывает. Гормональный профиль до наступления пубертатного периода неинформативен. После стимуляции ХГ уровень тестостерона бывает нормальный, при пробе на резистентность к андрогенам концентрация СССГ снижается в меньшей степени, чем в норме, в отличие от пациентов с полной резистентностью, у которых она не снижается (Sinnecker u. Kohler, 1989; Sinnecker et al., 1997). Различны также биохимические изменения рецепторов андрогенов, которые часто носят качественный характер. Однако нередко в исследованиях на культуре фибробластов, полученных из кожи половых органов, дефект рецепторов выявить не удается (Griffin, 1992). Анализ ДНК позволяет верифицировать диагноз, отличить наследственные формы от новых мутаций и выявить соматический мозаицизм (Hiort et al., 1993; Hiort et al, 1998; Holterhus et al., 1997).
От практиковавшегося ранее пробного лечения тестостероном детей, которых можно было воспитывать как девочек, в настоящее время отказались. У таких детей следует по возможности уменьшить риск дополнительной вирилизации во избежание гипертрофии клитора.
Лечение
Выбор пола для дальнейшего воспитания ребенка зависит от того, насколько выражена недостаточность маскулинизации. Чтобы оценить шансы на достижение достаточной вирилизации лечением высокими дозами тестостерона, проводят пробу на резистентность к андрогенам, а при семейной форме заболевания — также пробное лечение кого-нибудь из более старших членов семьи с этим заболеванием (Sinnecker et al., 1997). В пубертатном периоде часто развивается выраженная гинекомастия. Если предполагается слабый ответ на лечение тестостероном, даже при высоких его дозах, ребенка с амбивалентными половыми органами следует воспитывать как девочку. Для этого по возможности раньше (в течение первых 2 лет жизни) прибегают к феминизации наружных половых органов с помощью пластики вульвы. Пластику преддверия влагалища и самого влагалища выполняют в конце пубертатного периода или в более поздние сроки (см. ниже). Гонадэктомию следует выполнять по двум соображениям: во-первых, из-за высокого риска злокачественного перерождения, во-вторых, из-за возможности вирилизации в пубертатном периоде. В отличие от полной резистентности к андрогенам, при частичной резистентности яички у детей, которые воспитываются как девочки, следует обязательно удалять до наступления пубертатного периода. В пубертатном периоде проводят заместительную терапию эстрогенами и гестагенами.
Ложный женский гермафродитизм
Определение
Лица с ложным женским гермафродитизмом имеют генетически детерминированный женский пол (TDF отсутствует), яичники и развитые по женскому типу внутренние половые органы, а также вирилизированные наруэкные половые органы.
Ложный женский гермафродитизм характеризуется разнообразием клинических проявлений: от женского фенотипа и легкой вирилизации половых органов, множества вариантов амбивалентных половых органов до внешне нормально развитых мужских гениталий с «пустой» мошонкой.
Поскольку развитие внутренних и наружных половых органов при отсутствии функционирующих яичек всегда происходит по женскому типу, то вирилизация плода женского пола может произойти лишь при условии экстрагонадного происхождения андрогенов. Источником андрогенов может быть сам плод (АГС) или мать (прием препаратов, опухоли, лютеома беременности). Механизм развития ложного женского гермафродитизма показан на рис. 7-21.
Адреногенитольный синдром
Наиболее частой причиной ложного женского гермафродитизма является недостаточность фермента CYP21 (21-гидроксилаза). Этот фермент локализуется в корковом веществе надпочечников и катализирует превращение 17в-гидроксипрогесте- рона в кортизол и прогестерона в альдостерон. Накопление субстрата (17в-гидроксипрогестерон), обусловленное недостаточностью фермента, приводит к повышенному образованию андростендиона и тестостерона, которое влечет за собой вирилизацию наружных половых органов. Клиническая картина в зависимости от степени недостаточности 21-гидроксилазы включает симптомы, характерные для избытка андрогенов и недостаточности кортизола, реже — альдостерона.

Рис. 7-21. Механизм развитая ложного женского гермафродитазма (пораженная область при тех или иных нарушениях окрашена в красный цвет или затушевана).
Степень вирилизации оценивают по классификации Прадера (рис. 7-22).
Недостаточность фермента CYP11B1 (11в-гидроксилаза) приводит к нарушению превращения 11- дезоксикортизола в кортизол и 11 -дезоксикортикостерона в кортикостерон. При этом нарушении также отмечается избыточная продукция андрогенов, приводящая к внутриутробной вирилизации плода женского пола (рис. 7-23).
Недостаточность плацентарной ароматазы
Плацентарная ароматаза CYP19 катализирует превращение андростендиона и 16в-гидроксиандро- стендиона в эстрогены, предохраняя таким образом плод от вирилизирующего влияния плацентарных андрогенов. При недостаточности ароматазы не только нарушается синтез эстрогенов, но и происходит накопление андростендиона и тестостерона, которое вызывает внутриутробную вирилизацию плода женского пола (Dittmann, 1989; Shozu et al, 1991).
У девочек при рождении отмечаются выраженная в той или иной степени вирилизация наружных половых органов и нормально развитые по женскому типу внутренние половые органы. Для пубертатного периода характерны гипергонадотропный гипогонадизм, гиперандрогенемия и поликистозные яички. Созревание костной ткани (но не рост) замедлено, и нарушена минерализация костей. Эти проявления можно устранить заместительной терапией эстрогенами.
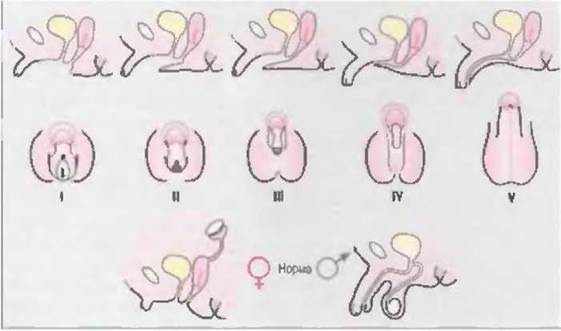
Рис. 7-22. Кпассификация фенотипов при ложном женском гермафродитизме (Prader, 1978).
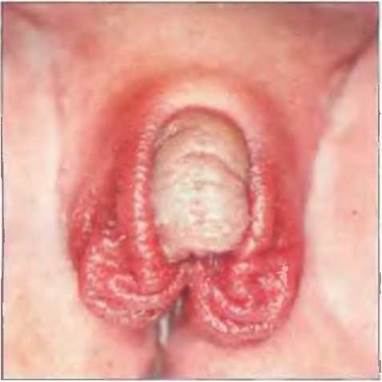
Рис. 7-23. Пациентка с генетически детерминированным женским полом (кариотип 46, XX) с ложным женским гермафродита - мом при АГС.
Трансплацентарная вирилизация
Экзогенные материнские андрогены
Назначение беременным производных тестостерона и гестагенов (в том числе производных 19- нортестостерона) может вызвать внутриутробную вирилизацию плода женского пола (Grum-bach et al., 1959). Степень вирилизации зависит от вида стероидного препарата и его дозы и особенно от сроков приема препаратов. Если он приходится на период после 12-й недели после зачатия, то сращение губно-мошоночных складок уже не произойдет, а разовьется только гипертрофия клитора.
Эндогенные материнские андрогены
Источником андрогенов, поступающих от матери и вызывающих внутриутробную вирилизацию плода женского пола, могут быть также вирилизирующие опухоли надпочечников или яичников (Murset et al., 1970). Образование андрогенов, вызывающих вирилизацию плода, может быть также обусловлено АГС у матери, лютеомой беременности и лютеиновыми кистами яичников (Malinak u. Miller, 1965). Как и при недостаточности плацентарной ароматазы. диагностическое значение имеет вирилизация матери во время беременности.
Необходимость в специальном лечении обычно отсутствует, так как после рождения вирилизация не прогрессирует, а в пубертатном периоде появляются женские вторичные половые признаки. К хирургической коррекции половых органов прибегают лишь в тех случаях, когда маскулинизация их сильно выражена. Если она заключается только в гипертрофии клитора, то по мере роста ребенка размеры клитора могут постепенно уменьшиться (собственное наблюдение). Поэтому при гипертрофии клитора хирургическая коррекция оправдана у тех пациенток, у которых клитор по размерам напоминает половой член.
Истинный гермафродитизм
Определение
Истинный гермафродитизм характеризуется наличием у пациентов одновременно яичников и яичек.
Чаще у пациентов на одной стороне имеется половая железа смешанного строения (овотестис), а на другой — яичник или яичко (реже). Однако возможны и другие комбинации, например яичник на одной стороне и яичко на другой или овотестис с обеих сторон.
Этиология и патогенез
Причинами истинного гермафродитизма могут быть (рис. 7-24):
• «мозаицизм по половым хромосомам;
• транслокации Y-хромосомы или ее фрагментов на аутосомы или Х-хромосомы;
• химеризм.
Лица с истинным гермафродитизмом обычно имеют кариотип 46,XX, реже — 46,XY или мозаицизм по половым хромосомам. Считают, что при «скрытом» мозаицизме происходит транслокация генов Y-хромосомы (ген SRY) на аутосомы или половые хромосомы. Кроме того, при двойном оплодотворении или слиянии двух нормальных оплодотворенных яйцеклеток возникает химеризм 46,XX/46,XY (Erhardt, 1979).
Клиническая картина
Клиническая картина характеризуется значительным разнообразием. Половые органы обычно развиты по промежуточному типу (амбивалентные), однако могут быть развиты также преимущественно по женскому или мужскому типу. Частыми симптомами являются гипоспадия, неполное сращение губно-мошоночных складок, крипторхизм и паховые грыжи, в которых обнаруживают гонады или производные мюллеровых протоков. Развитие внутренних половых органов соответствует ипсилатеральной гонаде: на стороне расположения яичка происходит стимуляция вольфова протока и подавление мюллерова, а на стороне расположения яичника развиваются производные мюллерова протока, в то время как вольфов проток подвергается обратному развитию. Поскольку тестикулярная часть овотестиса обычно бывает дисгенетичной, на его стороне внутренние половые органы развиваются по женскому типу. В пубертатном периоде происходит частичная вирилизация и развивается гинекомастия, часто появляются менструации.

Рис. 7-24. Механизм возникновения истинного гермафродитизма
Диагностика
Развитие половых органов по промежуточному типу и пальпируемый овотестис позволяют поставить предварительный диагноз истинного гермафродитизма. Диагностическое значение имеют кариотип 46,XX/46,XY, стимуляционная проба с помощью ХГ и менопаузального гормона, подтверждающая наличие как тестикулярной, так и овариальной ткани, а также гистологическое исследование половых желез, при котором обнаруживают оба вида ткани.
Лечение
Выбор пола определяется анатомическими соотношениями и возможностью хирургической коррекции. У детей, которых воспитывают как девочек, следует по возможности раньше удалить тестикулярную ткань из-за высокого риска ее злокачественного перерождения и профилактики вирилизации. У них возможно нормальное течение пубертатного периода, так как функция яичника может оказаться для этого достаточной (Nihoul- Fekete et al., 1984). У детей, которых воспитывают как мальчиков, следует удалить производные мюллеровых протоков и овариальную ткань. Поскольку тестикулярная часть овотестиса часто дисгенетична, то ее также следует удалить. Яички, расположенные в мошонке, сохраняют, но при этом регулярно следят за их состоянием. В отдельных случаях при необходимости проводят заместительную гормональную терапию и/или выполняют корригирующую операцию.
7.4. Тактика лечения при гермафродитизме
Определение пола
Быстрый и обоснованный выбор пола крайне важен для дальнейшего развития ребенка с амбивалентными гениталиями. Этот выбор основывается на оценке того, при каком поле следует рассчитывать на нормальную функцию половых органов. Давая такую оценку, упитывают анатомические соотношения и особенно размеры полового члена и возможность его роста. Следует учитывать также возможности хирургической коррекции и уровень культуры семьи ребенка. Поэтому решение о поле ребенка принимается совместно несколькими специалистами — гинекологом, уфологом, детским хируфгом, психологом. генетиком.
Хирургическое лечение
Относительно хирургического лечения на сегодняшний день существуют различные мнения и даются разные рекомендации. Так, одни специалисты считают необходимой по возможности раннюю «полную коррекцию» как наружных, так и внутренних половых органов (пластика вульвы, влагалища, редукция клитора), другие, в частности члены «рабочей группы против насилия в педиатрии и гинекологии», выступают за отказ от пола как признака, характеризующего личность, и любого оперативного вмешательства у детей, так как дети еще не способны выразить свое отношение к этому вопросу.
К сожалению, как правило, невозможно знать, смогут ли маленькие дети и их родители ориентироваться в нашем двуполом обществе, если половая принадлежность ребенка однозначно не определена. По-прежнему все, что касается сексуальности, в значительной степени табуизировано. Поэтому родители, которым необходимо принять эту необычность своего ребенка и общаться с ним. ничего не скрывая, оказываются перед большой проблемой. Ребенок, принадлежащий к «третьему полу» (или не имеющий пола), в реальной жизни не только будет подвержен частым конфликтам с обществом, но может оказаться не принятым своими родителями и тем самым недостаточно защищенным.
Мы понимаем и одобряем тех, кто ратует за толерантность в обществе, за то, чтобы в нем с уважением, с христианской терпимостью относились к человеку независимо от происхождения, цвета кожи, расовой и половой принадлежности. К сожалению, мы не видим такой терпимости ни в нашем обществе, ни в друом социокультурном окружении. Достаточно у ребенка небольшого отклонения от нормы (малый рост, ожирение, плохая успеваемость, высокая одаренность, телесный дефект), чтобы его выделили в школьном коллективе и в группах досуа. Особенно травмируются дети с неопределенной половой принадлежностью в пубертатном периоде. Поэтому ничего не предпринимая, врач не снимает с себя ответственность за судьбу таких детей.
Необходимо после тщательного обследования и установления диагноза провести беседу с родителями ребенка и вместе с психологом, имеющим достаточный опыт в этой области, разработать индивидуальный план лечения.
Мы считаем, что наружные половые органы, если это необходимо, нужно реконструировать в течение первого, но не позднее второго года жизни таким образом, чтобы они полностью соответствовали женским (пластика преддверия влагалища). Это важно для того, чтобы как сам ребенок, так и его сверстники не усомнились в его принадлежности к женском) полу. Редукционную пластику клитора, при которой удаляют лишь его «ствол», сохраняя сосудисто-нервные пучки, в этом возрасте предпринимают лишь в том случае, если размеры клитора слишком велик и он напоминает половой член. Поскольку отдаленные результаты таких операций еще не изучены, отношение к ним должно быть сдержанным. Ранее практиковавшаяся клиторэктомия в настоящее время не применяется. Вагинопластику или операцию по созданию нового влагалища у детей не выполняют, чтобы избежать травмирующих переживаний и болезненного послеоперационного долечивания (например, бужирования). Исходя из сказанного, объем хирургической коррекции наружных половых органов у детей должен быть минимальным.
В период полового созревания у пациентки может сформироваться осознанное желание начать половую жизнь. К этому желанию следует отнестись с уважением, оно также должно послужить обоснованием к хирургической коррекции. Пациентке не следует жить с постоянным половым партнером, хотя при ее неуверенности в себе, которую порождает ее необычность, поиск полового партнера затруднителен, с другой стороны, это освобождает ее от обязательств. С наступлением пубертатного периода ткани половых органов под влиянием эстрогенов становятся более податливыми и эластичными. Пациентки уже могут самостоятельно принимать решения, касающиеся изменения своего тела и выполнения хирургической коррекции. Лишь к этому времени должны выполняться большие по объему реконструктивные вмешательства с учетом пожелания пациенток, а не только их родителей.
У лиц с генетически детерминированным мужским полом (наличие Y-хромосомы), которые воспитываются как девочки, необходимо раннее удаление гонад (до начала пубертатного периода). Остаточная функция гонад таит в себе риск гетеросексуального (мужского) развития в пубертатном периоде. Единственным исключением являются случаи полной резистентности к андрогенам, при которой гонады следует сохранять и после пубертатного периода, чтобы способствовать спонтанной феминизации. В связи с риском злокачественного перерождения дисгенетичные гонады, клетки которых содержат Y-хромосому, подлежат удалению также у пациентов, которые воспитываются как мальчики. Лишь при условии, что яички находятся в мошонке, отчетливо пальпируются и имеется возможность наблюдать пациентов, яички оставляют, так как при достаточной остаточной функции они могли обеспечить спонтанное половое созревание мальчиков.
Гормональная терапия
Заместительную терапию половыми гормонами у детей с гипогонадизмом начинают к началу пубертатного периода: у девочек в 12 лет, у мальчиков в 13 лет. Цель гормональной терапии — обеспечить половое развитие, близкое к нормальному.
Девочкам вначале назначают конъюгированные эстрогенные препараты по 0,3 мг/сут в течение длительного времени. Через 6—12 мес или после появления менструаций переходят на циклическую эстроген-гестагенную терапию. Доза препаратов при этом соответствует дозе при заместительной терапии. Эстрогенный препарат назначают с 1-ro по 21-й день, гестагенный (например, медроксипрогестерона ацетат) — с 12-го по 21-й, в период с 22-го по 28-й день препараты отменяют. Дозу эстрогенов в течение первых 2—3 лет повышают до соответствующей 0,6—1,25 мг конъюгированного препарата. Применение препаратов, подавляющих овуляцию, нецелесообразно из-за их сильного нефизиологичного действия.
У пациенток с задержкой роста (например, при синдроме Ульриха—Тернера) при назначении высоких доз эстрогенов следует учитывать их унетающее действие на рост тела.
Психологический аспект лечения
Рождение ребенка с амбивалентными гениталиями вызывает серьезные психосоциальные проблемы. Не следует пытаться определить половую принадлежность такого ребенка уже в родильном зале.
Не следует делать также предположений. Родителям необходимо объяснить, что пол ребенка пока точно установить невозможно. Им следует также сообщить, что формирование пола еще не завершено, так же как оказывается незавершенным иногда формирование сердца или почек. С помощью целенаправленного обследования следует по возможности быстро установить причину аномалии и связанные с ней анатомические изменения. И лишь после этого решением консилиума в составе детского эндокринолога, гинеколога, уролога (или детского хирурга), психолога и генетика с учетом психосоциального и культурного окружения ребенка и мнения родителей определяют его пол.
Конечно, такое решение не может быть абсолютно правильным при любых обстоятельствах. Однако следует приложить все усилия для того, чтобы правильно оценить каждый случай с учетом накопленных знаний и опыта и принять такое решение, которое в дальнейшем по возможности уменьшит проблемы ребенка. Мы считаем, что в этом вопросе следует проявлять сдержанность и осторожность и не одобряем радикальную коррекцию, которая направлена только на исправление анатомического дефекта без учета наклонностей ребенка, его характера и физиологических особенностей.
Однако при всей сдержанности и недостаточной изученности проблемы не следует доходить до отрицания необходимости лечения и оставлять ребенка и его родителей без помощи и поддержки.
Быстрое и решительное определение пола ребенка и постоянная врачебная и психологическая помощь, оказываемая специалистами смежных дисциплин, могут создать предпосылки для нормальной жизни (в том числе половой) ребенка в дальнейшем.
Литература
Andersson S., Barman DM., Jenkins E.P., Russell DIF. Deletion of steroid 5a-reductase 2 gene in male pseudohermaphroditism//Nature. - 1991. - Vol. 354. -P. 159-161.
Baker T. G. A quantitative and cytological study of genn cells in the human ovaries // Proc. R. Soc. Lond. В Biol. Sci. - 1963. - Vol. 158.-P.417.
Bose H.S., Sujiwara T., Strauss J.F. 3rd, Miller W.L. The pathophysiology and genetics of congenital lipiod adrenal hyperplasia. International Congenital Lipoid Adrenal Hyperplasia Consortium // N. Engl. J. Med. — 1996. — Vol. 335. - P. 1870-1878.
Brown T.K., Lubahn D.B., Wilson EM. et al. Functional characterization of naturally occurring mutual androgen receptors from subjects with complete androgen insensitivity// Mol. Endocrinol. - 1990. - Vol. 4. - P. 1759-1772.
Burstein S., Grumbach MM, Kaplan S.L Early detenni- nation of androgen-responsiveness is important in the management of microphallus // Lancet. — 1979. — Vol. 2. — P. 983-986.
Chung B., Picado-Leonard J., Напій M. et al. Cytochrome P-450cl7 (steroid 17a-hydroxylasc/l 7,20-lyasc): Cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues // Proc. Natl. Acad. Sci. USA. - 1987. - Vol. 84. -P. 407-311.
Conte FA., Grumbach MM, Ito Y. et al. A syndrome of female pseudohennaphroditism, hypergonadotropic hypogonadism, and multicystic ovaries associated with missense mutations in the gene encoding aromatase (P450arom) // J. Clin. Endocrinol. Metab. - 1994. - Vol. 78. - P. 1287— 1292.
Dittmann R. W. Pranatal wirksame Honnone und Verh- altensmerkmale von Patientinnen mil den beiden klassis- chen Varianten des 21-Hydroxylase-Defektes. Bin Beitrag zur Psychocndokrinologic des adrenogenitalen Syndroms. Europaische Hochschulschriften, Reihe VI Psychologic — Frankfurt; Bern; N.Y.; Paris: Peter Lang, 1989. - S. 1-333.
Ehrhardt Л.А. Psychosexual adjustment in adolescence in patients with congenital abnonnalities of their sex organs // Genetic Mechanisms of Sexual Development / Eds H.L. Vallet. I.H. Porter. - N.Y.: Academic Press. 1979. -P. 473- 483.
Ehrhardt AA., Epstein R., Money J. Fetal androgens and female gender identity in the early-treated adrenogenital syndrome // Johns Hopkins Med. J. — 1968. — Vol. 122. -P. 160-167.
Ehrhardt AA., Meyer-Bahlburg H.F.L. Effects of prenatal sex honnones on gender-related behavior // Science. —1981. -Vol. 211. -P. 1312-1318.
Faiman C., Winter J.S.D. The control of gonadotropin secretion in complete testicular feminization // J. Clia Endocrinol. Metab. -1974. - Vol. 39. -P. 631-638.
FakhryJ, KhourvÂ., Kotml P.S., Noto RA. Sonography of autonomous follicular ovarian cjsts in precocious pseudopuberty // J. Ultrasound. Med. - 1988. - Vol. 7. -P. 597-603.
FergusonSmith MA. Karyotype-phenotype correlations in gonadal dysgenesis and their bearing on the pathogenesis of malformations // J. Med. Genet. — 1965. — Vol. 2. — P. 142-155.
Ferguson-Smith MA., Goodfellow P.N SRY and primary' sex reversal syndromes // The Metabolic and Molecular Basis of Inherited Disease / Eds C.R. Scriver, A.L. Beau- det, W.S. Sly, D. Valle. - 7th ed. - N.Y.: McGraw-Hill,1995. -P. 739-748.
Griffin JE. Androgen resistance — the clinical and molecular spectrum // N. Engl. J. Med. - 1992. - Vol. 326. -P. 611-618.
Grumbach MM, Conte FA. Disorders of sex differentiation // Williams Textbook of Endocrinology / Eds J.D. Wilson, D.W. Foster, H.M. Kronenberg, P.R. Larsea — 9th ed. - Philadelphia: WB Saunders, 1998. - P. 1303-1425.
Grumbach MM., Ducharme J.R., Moloshok RE. On die fetal masculinizing action of certain oral progestins // J. Clin. Endocrinol. Metab. - 1959. - Vol. 19. - P. 1369-1380.
Hall P. F. Role of cytochromes P-450 in the biosynthesis of steroid hormones // Vitam. Honn. — 1985. — Vol. 42. -P. 315-368.
Hiort 0., Huang O., Sinnecker G.H. G. et al. Rapid characterization of mutations of the androgen receptor in patients with androgen insensitivity' syndromes: application for diagnosis, genetic counseling, and therapy // J. Clia Endocrinol. Metab. - 1993. - Vol. 77. - P. 262-266.
Hiort O, Sinnecker G.H.G., Holterhus PM. et al. The clinical and molecular spectrum of androgen insensitivity' syndromes // Am. J. Med. Genet. - 1996. - Vol. 63. - P. 218-222.
Hiort ()., Sinnecker G.H.G., Holterhus PM. et al. Inherited and de novo androgen receptor gene mutations: investigation of single case families // J. Pediatr. — 1998. — Vol. 132. -P. 939-943.
Hiort ()., Willenbring II., Albers N. et al. Molecular genetic analysis and hCG stimulation tests in the diagnosis of prepubertal patients with partial 5a-reductase deficiency' // Eur. J. Pediatr. - 1996. - Vol. 155. - P. 445-451.
Holterhus PM, Bruggenwirth H.T., Hiort O. et al. Mosaicism due to a somatic mutation of the androgen receptor gene determines phenotype in androgen insensitivity' syndrome // J. Clin. Endocrinol. Metab. — 1997. — Vol. 82. - P. 3584-3589.
Imperato-McGinley J., Guerrero L, Gautier T, Peterson R.E. Steroid 5a-reductase deficiency in man: an inherited form of male pseudohermaphroditism. — Science. — 1974. — Vol. 186. -P. 1213-1215.
Imperato-McGinley J., Peterson R.E., Gautier T, SturlaE. Male pseudohermaphroditism secondary' to 5a-reduc-tase deficiency — a model for the role of androgens in both the development of the male phenotype and the evolution of a male gender identity' // J. Steroid Biochem. — 1979. — Vol. 11. -P. 637-645.
Jasso N. L'honnone anti-mullerienne: une foeto-pro- teine? // Arch. Fr. Pediatr. - 1975. - Vol. 32. - P. 109-111.
Leshin M, Griffin J.E'., Wilson J.D. Hereditary' male pseudohermaphroditism associated with an unstable form of 5a-reductase // J. Clin. Invest. - 1978. - Vol. 62. - P. 685- 691.
MacDonaldP.C., Madden J.D., Brenner P.F. et al. Origin of estrogen in nonnal men and in women with testicular feminization//J. Clin. Endocrinol. Metab. — 1979. — Vol. 49. - P. 905-916.
Madan K., Gooren I.., Schoemaker J. Three cases of sex chromosome mosaicism with a nonfluorescent Y // Hum. Genet. - 1979. - Vol. 46. - P. 295-304.
Malinak L.R., Miller G. Г. Bilateral multicentric ovarian luteomas of pregnancy' associated with masculinization of a female infant // Am. J. Obstet. Gynecol. — 1965. -Vol. 91. -P. 251-256.
Manuel M, Katayama K.P., Jones H. IF. The age of occurrence of gonadal tumors in intersex patients with a Y chromosome // Ibid. - 1976. - Vol. 124. - P. 293-300.
Marcelli M, Tilley W.D., Wilson CM. et al. A single nucleotide substitution introduces a premature termination codon into the androgen receptor gene of a patient with receptor-negative androgen resistance // J. Clin. Invest. — 1990. - Vol. 85. - P. 1522-1528.
Masica D.N., Money J., Ehrhardt A A. Fetal feminization and female gender identity' in the testicular feminizing syndrome of androgen insensitivity // Arch. Sex. Beliav. — 1971. -Vol. 1. -P. 131-142.
McPhaul M.J., Marcelli M, Tilley W.D. et al. Molecular basis of androgen resistance in a family with a qualitative abnormality' of the androgen receptor and responsive to high- dose therapy // J. Clia Invest. - 1991. - Vol. 87. -P. 1413- 1421.
Money J., Ehrhardt .1.1. Man and Woman. Boy and Ggirl. — Baltimore; Lond.: The Johns Hopkins University' Press, 1972.
Money J., Schwartz M. Dating, romantic and nonromantic friendships, and sexuality in 17 early-treated adrenogenital females, aged 16—25 // Congenital Adrenal Hyperplasia / Eds P.A. Lee, L.P. Plotnick, A.A. Kowarski, C.J. Migeoa — Baltimore: University' Park Press, 1977. — P. 419-431.
Money J., Schwartz M, Lewis EG. Adult erotosexual status and fetal hormonal masculinization and demasculin- ization: 46,XX congenital virilizing adrenal hyperplasia and 46,XY androgen-insensitivity syndrome compared // Psychoneuroendocrinology. - 1984. - Vol. 9. -P. 405-414.
Moore R.J., Griffin J.E., Wilson J.D. Diminished 5a- reductase activity' in extracts of fibroblasts cultured from patients with familial incomplete male pseudohennaphroditism. type 2 // J. Biol. Chem. - 1975. - Vol. 250. - P. 7168-7172.
Mueller R.F. The Denys-Drash syndrome // J. Med. Genet. - 1994. - Vol. 31. - P. 471-177.
Muller M, Bidlingmeier R, Forster C, Knorr D. Psychosexuelles Verhalten von Frauen mil adrenogenitalem Syndrom // Helv. Paediatr. Acta. - 1982. - Bd 37. - S. 571- 580.
Mur set G, Zaclmiann M, Prader A. et al. Male external genitalia of a girl caused by a virilizing adrenal tumor in the mother. Case report and steroid studies // Acta Endocrinol. (Copenli.). - 1970. - Vol. 65. - P. 627-638.
Nihoul-Fekete C, Lortat-Jacob S., Cochin ()., Josso N. Preservation of gonadal function in tme hennaphroditism // J. Pediatr. Surg. - 1984. - Vol. 19. - P. 50-55.
Nowakowski IL, Lenz W Genetic aspects in male hypogonadism // Recent Prog. Honn. Res. — 1961. — Vol. 17. -P. 53.
O'Rahilly. The development of the vagina in the human // Birth Defects Orig. Artie. Ser. - 1977. - Vol. 13. -P. 123- 136.
Pinsky F, Kaufinan M, Straisfeld C. et al. 5a-reductase activity of genital and nongenital skin fibroblasts from patients with 5a-reductase deficiency, androgen insensitivity, or unknown fonns of male pseudohennaphroditism // Am. J. Med. Genet. - 1978. - Vol. 1. - P. 407-416.
Prader A. Storungen der Geschlechtsdifferenzierung (In- tersexualitat) // Klinik der inneren Sekretion / Labhardt A. (Hrsg). — Berlin; Heidelberg; N.Y.: Springer, 1978. -S. 654-688.
Price P., Wass JA.H., Griffin J.E. et al. High dose androgen therapy in male pseudohennaphroditism due to 5a- reductase deficiency' and disorders of the androgen receptor // J. Clia Invest. - 1984. - Vol. 74. - P. 1496-1508.
Rubin R.T., Reinisch JM, HaskettR.F. Postnatal gonadal steroid effects on human behavior // Science. — 1981. - Vol. 211.-P. 1318-1324.
Rutgers J.F, Scully R.E. The androgen insensitivity syndrome (testicular feminization): a clinicopathologic study of 43 cases // Int. J. Gynaecol. Pathol. -1991. - Vol. 10. -P. 126-144.
Shozu M, Akasofu K., Harada T, Kubota Y. A new cause of female pseudohermaphroditism: placental aro-matase deficiency // J. Clin. Endocrinol. Metab. — 1991. — Vol. 72. - P. 560-566.
Siiteri P.K., Wilson J.D. Testosterone fonnation and metabolism during male sexual differentiation in the human embryo // Ibid. - 1974. - Vol. 38. - P. 113-125.
Sinclair A.H., Berta P., Palmer MS. et al. A gene from the human sex-detennining region encodes a protein with homology to a conserved DNA-binding motif// Nature. — 1990. - Vol. 346. - P. 240-244.
Sinnecker G., Kohler S. Sex honnone-binding globulin response to the anabolic steroid stanozolol: evidence for its suitability as a biological androgen sensitivity' test // J. Clin. Endocrinol. Metab. - 1989. - Vol. 68. -P. 1195-200.
Sinnecker G.H.G., Hiort O., Dihhelt L. et al. Phenotypic classification of male pseudohennaphroditism due to steroid 5a-reductase 2 deficiency // Am. J. Med. Genet. —1996. - Vol. 63. -P.223-230.
Sinnecker G.H.G, Hiort ()., Nitsche E.M. et al. Functional assessment and clinical classification of androgen sensitivity in patients with mutations of the androgen receptor gene. Gennan Collaborative Intersex Study' Group // Eur. J. Pediatr. - 1997. - Vol. 156. - P. 7-14.
Sinnecker G.H.G., Sinnecker R, Muhlenberg R, Hiort O. Gender assigmnent in patients with partial androgen insensitivity syndrome: significance of genotype and assessment of androgen receptor function (Abstract 23) // Honn. Res. - 1995. - Vol. 44. - Suppl. 1. - P. 6.
Sinnecker G., WilligRP., Stahnke N, Braendle W. 46,XY Reine Gonadendysgenesie (Swyer-Syndrom). Klinische und endokrinologische Befunde // Monatsschr. Kinderheilkd. —1982.-Bd 130.-S. 795-797.
Stocco D.M, Clark B.J. The role of the steroidogenic acute regulatoiy protein in steroidogenesis // Steroids. —1997. - Vol. 62. - P. 29-36.
Themmen A.P.N., Brunner H.G. Luteinizing honnone receptor mutations and sex differentiation // Eur. J. Endocrinol. - 1996. - Vol. 134. - P. 533-540.
Tint G.S., Salen G, Botta A.K. et al. Conelation of severity and outcome with plasma sterol levels in variants of the Smith-Lemli-Opitz syndrome // J. Pediatr. — 1995. — Vol. 127. - P. 82-87.
Tsuchiya K, Reijo R, Page D.C., Disteche CM. Gona- doblastoma: molecular definition of the susceptibility region on the Y chromosome // Am. J. Hum. Genet. — 1995.- Vol. 57. - P. 1400-1407.
Verp MS., Simpson J.L. Abnonnal sexual differentiation and neoplasia // Cancer Genet. Cytogenet. — 1987. — Vol. 25.-P. 191-218.
Wagner T, Wirth J, Meyer J. et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9 // Cell. - 1994. -Vol. 79. -P. 1111-1120.
Walsh P.C., Madden J.D., Harrod MJ. et al. Familial incomplete male pseudohennaphroditism, type 2. Decreased dihydrotestosterone fonnation in pseudovaginal perineoscrotal hypospadias // N. Engl. J. Med. - 1974. - Vol. 291. - P. 944-949.